Characterization Methods for Quantifying Non-Specific Adsorption on Sensor Surfaces: A Guide for Biomedical Researchers
Non-specific adsorption (NSA) presents a major challenge in biosensor development, adversely affecting sensitivity, selectivity, and reliability, particularly in complex matrices like blood, serum, and environmental samples.
Characterization Methods for Quantifying Non-Specific Adsorption on Sensor Surfaces: A Guide for Biomedical Researchers
Abstract
Non-specific adsorption (NSA) presents a major challenge in biosensor development, adversely affecting sensitivity, selectivity, and reliability, particularly in complex matrices like blood, serum, and environmental samples. This article provides a comprehensive overview of the foundational principles, methodological approaches, and advanced strategies for characterizing and quantifying NSA on sensor surfaces. Tailored for researchers, scientists, and drug development professionals, it explores the mechanisms of NSA, details a range of characterization techniques from electrochemical to optical methods, and discusses optimization and troubleshooting protocols. Furthermore, it presents a comparative analysis of validation frameworks and future perspectives, including high-throughput screening and machine learning, aiming to equip professionals with the knowledge to design robust, fouling-resistant biosensors for clinical and environmental monitoring.
Understanding Non-Specific Adsorption: Mechanisms and Impact on Biosensor Performance
Non-specific adsorption (NSA) represents a fundamental challenge in the development and deployment of reliable biosensors. This phenomenon describes the undesirable accumulation of molecules—such as proteins, cells, or other biomolecules—from complex samples onto biosensing interfaces, which occurs through mechanisms distinct from the intended specific biorecognition event [1]. The performance, reliability, and accuracy of biosensors in clinical diagnostics, environmental monitoring, and drug development are critically dependent on effectively managing NSA, as it directly compromises signal integrity by increasing background noise, reducing sensitivity, and potentially leading to false positives or negatives [1] [2].
The process of NSA is primarily governed by two distinct physical mechanisms: physisorption (physical adsorption) and chemisorption (chemical adsorption). Physisorption involves weak, reversible interactions that do not alter the chemical structure of the adsorbate, while chemisorption involves stronger, often irreversible chemical bond formation [3]. In biosensing applications, both processes can occur simultaneously or competitively, with their relative contributions determined by the chemical properties of the sensor surface, the nature of the surrounding biological fluid, and operational conditions [1] [4]. Understanding the distinction between these mechanisms is not merely academic; it directly informs the selection of characterization techniques and the design of effective surface passivation strategies to mitigate fouling. For researchers and drug development professionals, controlling NSA is essential for translating laboratory biosensor research into robust, commercially viable diagnostic devices capable of functioning in complex matrices like blood, serum, or milk [1].
Fundamental Mechanisms: Physisorption and Chemisorption
The accumulation of non-target molecules on a sensor surface proceeds through distinct pathways governed by different physical forces. A clear comprehension of these mechanisms is the first step toward developing effective antifouling strategies.
Physisorption: Physical Adsorption
Physisorption is an adsorption process driven by weak, reversible intermolecular forces between the adsorbate and the adsorbent surface. The chemical identity of the adsorbed molecule remains unchanged.
- Binding Forces and Energy: The process is mediated by van der Waals forces, dipole-dipole interactions, and occasionally hydrogen bonds. These are low-energy interactions, typically with binding energies below 100 kJ/mol [3]. The weakness of these forces makes physisorption a readily reversible process.
- Reversibility and Multi-layer Formation: A key characteristic of physisorption is its reversibility. Adsorbed molecules can often be desorbed by applying gentle stimuli such as a change in temperature, a reduction in partial pressure, or a strong surfactinated rinse [4] [3]. Furthermore, because the forces are long-range and non-specific, physisorption can lead to the formation of multiple molecular layers on the surface [3].
- Temperature Dependence: Physisorption is favored at lower temperatures. As the thermal energy of molecules increases with temperature, the adsorbed molecules are more likely to overcome the weak attractive forces and desorb from the surface [3] [5].
Chemisorption: Chemical Adsorption
Chemisorption is a form of adsorption that involves the formation of strong chemical bonds—covalent or ionic—between the adsorbate and the atoms on the adsorbent surface. This process fundamentally changes the electronic structure of the adsorbed molecules.
- Binding Forces and Energy: Chemisorption involves significant chemical bond formation, with binding energies in the range of 200–800 kJ/mol [3]. This high energy makes the process often irreversible under normal sensor operating conditions, as desorption would require breaking these strong chemical bonds [3].
- Specificity and Monolayer Formation: Unlike physisorption, chemisorption is highly specific and typically results in the formation of only a monomolecular layer (monolayer). The reaction can only occur where specific, compatible chemical sites are available on the surface [3].
- Role in Fouling and Catalysis: In the context of biosensor fouling, chemisorption can lead to permanent passivation of the surface. However, this same mechanism is crucial in the initial functionalization of biosensors, where bioreceptors (e.g., antibodies, aptamers) are deliberately and stably immobilized onto the transducer surface via covalent bonds [1] [6].
Table 1: Comparative Characteristics of Physisorption and Chemisorption.
| Characteristic | Physisorption | Chemisorption |
|---|---|---|
| Binding Force | Van der Waals, dipole-dipole | Covalent, ionic bonds |
| Binding Energy | Low (< 100 kJ/mol) | High (200-800 kJ/mol) |
| Reversibility | Highly reversible | Often irreversible |
| Adsorption Layers | Multi-layer | Mono-layer |
| Temperature Dependence | Favored at low temperatures | Often favored at higher temperatures |
| Specificity | Non-specific | Highly specific |
| Role in Biosensing | Primary source of NSA | Used for bioreceptor immobilization; can cause NSA |
The following diagram illustrates the fundamental differences in the interaction mechanisms and outcomes of physisorption and chemisorption on a sensor surface.
Quantitative Discrimination and Experimental Characterization
Discriminating between physisorption and chemisorption is critical for diagnosing the root cause of NSA and formulating an appropriate mitigation strategy. Researchers employ a suite of quantitative tools and experimental protocols to probe these interactions.
A Quantitative Discrimination Method
A seminal study by Yang et al. (2016) established a robust protocol for discriminating specific from non-specific lectin-glycan interactions on silicon surfaces, a methodology that can be adapted for general NSA analysis [4]. The core finding was that protein physisorption was more prevalent than specific chemisorption across common washing protocols. The study demonstrated that this physisorption could be effectively suppressed by applying a strong surfactinated rinse, which disrupts weak physical bonds without affecting covalent ones [4].
The experimental workflow involved:
- Surface Fabrication: Creating well-defined glycan (mannoside and lactoside) monolayers immobilized on hydrogenated crystalline silicon (111) surfaces using a "click" chemistry-based conjugation protocol to ensure controlled density [4].
- Protein Exposure: Incubating the functionalized surfaces with target lectins (Lens culinaris and Peanut agglutinin) [4].
- Quantitative Analysis: Using quantitative Fourier-Transform Infrared Spectroscopy in Attenuated Total Reflection mode (FTIR-ATR) to measure the amount of adsorbed protein. The data was interpreted using various adsorption isotherm models to quantify binding [4].
- Surface Imaging: Employing Atomic Force Microscopy (AFM) to visualize the distribution and morphology of the adsorbed proteins, corroborating the spectroscopic data [4].
The combination of quantitative FTIR and AFM provided a powerful correlation between the quantity of adsorbed protein and its physical distribution on the surface, conclusively demonstrating the coexistence of physisorption and chemisorption.
Key Characterization Techniques and Their Outputs
Different analytical techniques provide unique insights into the nature of adsorption events. The following table summarizes the primary methods used to characterize NSA and their capabilities in distinguishing between physisorption and chemisorption.
Table 2: Experimental Techniques for Characterizing Non-Specific Adsorption.
| Technique | Key Measurable Parameters | Utility for Physisorption | Utility for Chemisorption |
|---|---|---|---|
| Surface Plasmon Resonance (SPR) | Mass change on surface, binding kinetics (ka, kd), affinity (KD) [1]. | Detects rapid, reversible binding; signal often decreases with surfactant rinse. | Detects stable, irreversible binding; signal persists after harsh rinsing. |
| Electrochemical (EC) Methods | Electron transfer rate, charge transfer resistance, signal drift [1]. | Monitors passivation layer formation causing signal drift. | Can detect irreversible blocking of electroactive sites. |
| Quantitative FTIR-ATR | Chemical bond vibration, functional group identification, quantitative adsorbed amount [4]. | Identifies lack of new chemical bonds; quantifies weakly bound adsorbate. | Detects formation of new covalent bonds; quantifies strongly bound adsorbate. |
| Atomic Force Microscopy (AFM) | Surface topography, adhesion forces, nanoscale morphology [4]. | Visualizes diffuse or multi-layer coverage; measures weak adhesion forces. | Visualizes ordered mono-layers; measures strong, specific adhesion. |
| Adsorption Isotherm Analysis | Surface coverage (Θ), binding constant, monolayer capacity [3]. | Fits models for multi-layer adsorption (e.g., BET isotherm). | Fits models for monolayer adsorption on homogeneous sites (e.g., Langmuir isotherm). |
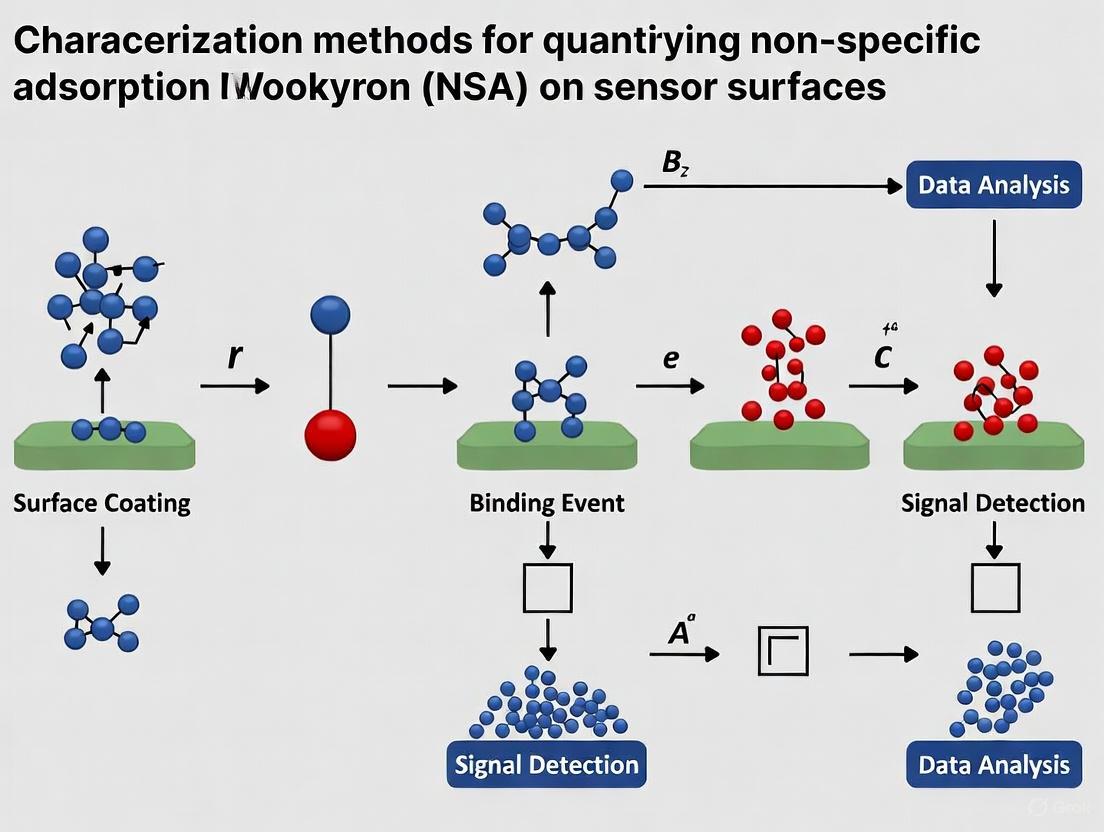
The process of characterizing NSA and identifying the dominant adsorption mechanism follows a logical experimental workflow, which can be designed as follows:
Promising Solutions: From Fundamentals to Applications
Addressing NSA requires strategies tailored to the dominant adsorption mechanism. Recent research has shifted from simple passive blocking methods to advanced surface engineering and active removal techniques.
Advanced Antifouling Coatings and Materials
The development of novel coatings that create a physical and energetic barrier to foulants is a primary strategy. These materials are designed to minimize both physisorption and chemisorption by presenting a surface that is inert and non-interactive.
- Zwitterionic Peptides: These have emerged as a superior alternative to polyethyleneglycol (PEG), the traditional "gold standard." A 2025 study by Awawdeh et al. demonstrated that a specific zwitterionic peptide with the sequence EKEKEKEKEKGGC, when covalently immobilized on porous silicon (PSi) biosensors, provided broad-spectrum protection against fouling from gastrointestinal fluid and bacterial lysate [7]. The peptide's repeating motifs of glutamic acid (E, negatively charged) and lysine (K, positively charged) create a net-neutral, hydrophilic surface that binds a tight hydration layer via electrostatic interactions, forming a formidable barrier against NSA [7]. The sensor functionalized with this peptide showed an order of magnitude improvement in the limit of detection and signal-to-noise ratio for lactoferrin detection compared to PEG-passivated sensors [7].
- Hybrid and Nanomaterial-Enhanced Coatings: The integration of nanomaterials like graphene, carbon nanotubes, and gold nanoparticles is revolutionizing surface functionalization. Their high surface-to-volume ratio and tunable optoelectronic properties allow for dense immobilization of bioreceptors and superior signal transduction [6]. Furthermore, metal-organic frameworks (MOFs) are being explored as nanozymes and porous scaffolds that can be functionalized with antifouling groups, enhancing both catalytic activity and selectivity in complex matrices [8].
The Role of Artificial Intelligence and Computational Design
A paradigm shift is underway with the integration of Artificial Intelligence (AI) and Machine Learning (ML) into biosensor development. AI-driven models are accelerating the rational design of antifouling interfaces by predicting optimal surface architectures and materials compositions without relying solely on trial-and-error experimentation [6].
- Machine Learning Optimization: ML algorithms analyze complex relationships between surface properties (e.g., hydrophobicity, charge distribution) and sensor performance metrics (e.g., limit of detection, nonspecific binding) to identify optimal functionalization strategies [6].
- Molecular Dynamics (MD) Simulations: AI-guided MD simulations provide atomic-level insights into the interactions between biomolecules and functionalized surfaces. This helps in understanding the fundamental mechanisms of fouling and in designing high-affinity binding surfaces or robust antifouling coatings [6].
- Density Functional Theory (DFT) for Material Design: Computational advances are also improving the accuracy of modeling adsorption itself. For instance, the development of the Opt(MS+rVV10) density functional aims to achieve chemical accuracy in predicting both chemisorption and physisorption energies, which is crucial for the in-silico design of new sensor materials with minimal NSA propensity [9].
Research Toolkit: Essential Reagents and Materials
The following table catalogs key reagents and materials essential for researchers developing and characterizing antifouling sensor interfaces.
Table 3: Essential Research Reagent Solutions for NSA Studies.
| Reagent/Material | Function/Application | Key Characteristic |
|---|---|---|
| Zwitterionic Peptides (e.g., EKEKEKEKEKGGC) [7] | Covalent surface passivation to prevent NSA of proteins and cells. | Forms a strong hydration layer; net-neutral charge; superior to PEG. |
| Polyethylene Glycol (PEG) [1] [7] | Traditional blocking agent for surface passivation. | Hydrophilic; forms a hydration barrier; prone to oxidative degradation. |
| Silane Coupling Agents (e.g., APTES) [6] | Creates a functional interface (e.g., amine groups) on oxide surfaces (SiO₂) for subsequent bioconjugation. | Enables covalent immobilization of bioreceptors or antifouling layers. |
| Alkanethiols [6] | Formation of Self-Assembled Monolayers (SAMs) on gold surfaces for controlled surface engineering. | Provides a well-defined, tunable surface chemistry for fundamental studies. |
| Surfactant Solutions (e.g., Tween 20) [4] | Used in rinse protocols to discriminate and remove physisorbed molecules. | Disrupts weak van der Waals and hydrophobic interactions. |
| Quantitative FTIR-ATR [4] | Technique to quantitatively measure the amount and nature of adsorbed species. | Provides chemical bond information and quantifies adsorption. |
| Functionalized Porous Silicon (PSi) [7] | High-surface-area transducer model for studying NSA in challenging porous structures. | Amplifies fouling challenges, making it a stringent testbed for antifouling strategies. |
| Gold Nanoparticles (AuNPs) [6] | Nanomaterial for signal amplification and enhanced bioreceptor immobilization on sensing interfaces. | High surface-to-volume ratio; tunable optic and electronic properties. |
The systematic discrimination between physisorption and chemisorption is a cornerstone of developing robust biosensors resistant to non-specific adsorption. While physisorption, governed by weak van der Waals forces, is often reversible and addressable via optimized rinsing protocols, chemisorption presents a more challenging problem due to its irreversible, covalent nature [4] [3]. The future of mitigating NSA lies in the rational design of advanced functional interfaces. The integration of novel materials like zwitterionic peptides [7] with computational and AI-driven design tools [6] [9] represents a powerful interdisciplinary approach. By moving beyond traditional trial-and-error methods, researchers can now predict and engineer surfaces with inherent antifouling properties, accelerating the development of highly reliable biosensors for critical applications in drug development and clinical diagnostics.
The characterization of interactions on sensor surfaces is a cornerstone of modern pharmaceutical and analytical sciences, particularly in the detection of non-steroidal anti-inflammatory drugs (NSAIDs). A comprehensive understanding of the primary molecular interactions—electrostatic, hydrophobic, and van der Waals—is crucial for developing sensitive, selective, and reliable sensing platforms. These fundamental forces govern the binding affinity, specificity, and overall performance of sensors, influencing their response to target analytes in complex matrices such as biological fluids and environmental samples. Within the broader thesis on characterization methods for quantifying NSAID interactions on sensor surfaces, this guide objectively compares how these three interaction mechanisms contribute to sensor performance across various technological platforms. We present experimental data and detailed methodologies to provide researchers, scientists, and drug development professionals with a practical framework for evaluating and selecting appropriate sensing strategies based on the dominant interaction forces they exploit.
Comparative Analysis of Primary Interaction Mechanisms
The table below summarizes the comparative role and performance of electrostatic, hydrophobic, and van der Waals interactions across various NSAID sensing and characterization platforms, synthesizing data from multiple experimental studies.
Table 1: Comparison of Primary Interaction Mechanisms in NSAID Sensing and Characterization
| Interaction Type | Role in NSAID Sensing/Characterization | Experimental Evidence & Performance Data | Key Amino Acids/Functional Groups Involved | Detection Range/ Sensitivity |
|---|---|---|---|---|
| Electrostatic | Dominant in COX enzyme selectivity [10]; Key for virtual screening (ES-Screen) [11]; Critical for carboxylate binding in optical sensors [12]. | Quantum crystallography revealed binding energy differences: Flurbiprofen (strongest), Celecoxib/Meloxicam (COX-2 selective) [10]; ES-Screen showed superior enrichment (AUC >0.75) vs. docking/GBSA/PBSA [11]. | Arg120, His513, Tyr355 in COX active site [10]; Carboxylate group of NSAIDs [12] [13]. | ES-Screen EF1% (Enrichment Factor at 1%) outperformed other methods for most of 53 protein targets [11]. |
| Hydrophobic | Contributes to COX binding affinity and selectivity [10]; Critical for drug-protein binding (e.g., HSA) [14]; Enhances sensor selectivity via non-polar cavities. | QSAR studies: NSAID activity highly correlated with lipophilicity (log P value) [15]; NMR identified aromatic moieties of Diclofenac, Ketorolac as key in HSA binding [14]. | Aromatic moieties of drugs (e.g., Diclofenac, Ketorolac) [14]; Left flipper/dorsal fin domains in P2X3R [13]. | Calculated log P values directly correlated with anti-inflammatory activity in QSAR models [15]. |
| van der Waals | Provides structural complementarity in protein-ligand complexes [10]; Contributes to binding stability in sensor-analyte interfaces; Important for non-polar replacement energies in virtual screening [11]. | ES-Screen integrates van der Waals solvation energies for molecular discrimination [11]; Contributes to stabilization in protein-ligand complexes analyzed via quantum crystallography [10]. | Various binding site residues enabling shape complementarity [10] [11]. | ES-Screen performance improved by integrating van der Waals replacement energies with electrostatic terms [11]. |
Experimental Protocols for Characterizing Interaction Mechanisms
Electrostatic Interaction Characterization
Protocol 1: Electrostatic Potential Mapping for Virtual Screening (ES-Screen Method)
The ES-Screen method provides a unique electrostatics-driven approach for virtual screening, independent of molecular docking [11].
- Principle: Calculates the energy cost of replacing a cognate ligand with a query molecule, focusing on electrostatic replacement energies. This optimizes for thermodynamic stability relative to a known stable binding state.
- Procedure:
- Ligand Pose Generation: Input initial ligand poses using knowledge-based pharmacophore models derived from protein-ligand crystal structures, refined with excluded volume spheres to minimize atomic clashes.
- Electrostatic Potential (ESP) Extrapolation: Extrapolate the ligand-free protein electrostatic potential to atom positions within the binding site.
- Replacement Energy Calculation: Assemble ligand atom partial charges from solvent (high dielectric) into the binding site (low dielectric). Calculate single-point electrostatic interaction energies for both reference (cognate) and query ligands.
- Scoring and Ranking: Calculate electrostatic replacement energy as the difference between query and reference ligand energies. Normalize and combine with shape and physicochemical similarity terms to generate a Z-score for ranking.
- Key Reagents & Parameters: Protein-ligand co-crystal structure (reference), query ligand library, dielectric constants for solvent and binding site, partial charge assignments, shape similarity metrics.
Table 2: Research Reagent Solutions for Interaction Characterization
| Reagent/Material | Function/Application | Example Use Case |
|---|---|---|
| Cadmium Sulfide (CdS) Nanoparticles | Electrode modifier; enhances electrocatalytic signal via surface interactions. | 4-fold enhancement in naproxen electro-oxidation signals vs. bare GCE [16]. |
| Zinc Oxide (ZnO) Nanoparticles | Electrode modifier; biocompatible semiconductor for signal amplification. | 2-fold enhancement in mobic electro-oxidation signals [16]. |
| Human Serum Albumin (HSA) | Model transport protein for studying drug-protein binding interactions. | NMR investigation of Diclofenac, Ketorolac, Flurbiprofen binding affinity and sites [14]. |
| Molecularly Imprinted Polymers (MIPs) | Synthetic receptors with tailored cavities for selective analyte binding. | Pretreatment and sensing of NSAIDs via complementary shape and functional groups [17]. |
| UBDB Database + EPMM Method | Transferable aspherical pseudoatoms database for accurate electrostatic interaction energy calculation. | Quantum crystallography analysis of NSAID binding to COX enzymes [10]. |
Hydrophobic Interaction Characterization
Protocol 2: NMR Spectroscopy for Drug-Protein Binding Affinity
Solution Nuclear Magnetic Resonance (NMR) spectroscopy is a powerful tool for investigating interaction strength and identifying binding sites, particularly hydrophobic moieties.
- Principle: Measures changes in relaxation parameters of drug protons when bound to a macromolecule like Human Serum Albumin (HSA). The normalized affinity index quantifies interaction strength.
- Procedure: [14]
- Sample Preparation: Prepare drug solutions in D₂O buffer (e.g., pH 7.4 phosphate buffer) with increasing concentrations of HSA.
- Relaxation Rate Measurement: For each drug/HSA mixture, measure mono-selective spin-lattice relaxation times (T₁ₘₛ) for resolved drug proton signals.
- Data Analysis: Calculate mono-selective relaxation rates (R₁ₘₛ = 1/T₁ₘₛ). Plot normalized relaxation rates (ΔR/Rf) against HSA concentration.
- Affinity Index Calculation: The slope of the linear fit yields the normalized affinity index ([Aₙ]), a measure of global drug-HSA affinity. Higher values indicate stronger binding.
- Key Findings: Aromatic protons and those near carboxylate groups show the largest relaxation rate increases, indicating their central role in binding, often via hydrophobic effects and van der Waals contacts [14].
van der Waals and Combined Interaction Analysis
Protocol 3: Quantum Crystallography for Protein-Ligand Interaction Energetics
This advanced method combines high-resolution X-ray crystallography with transferable aspherical atom models to elucidate comprehensive interaction profiles.
- Principle: Uses the transferable aspherical pseudoatom databank (UBDB) and the Exact Potential/Multipole Model (EPMM) to compute highly accurate electrostatic, van der Waals, and other interaction energies from crystallographic data.
- Procedure: [10]
- Data Collection: Obtain high-resolution X-ray diffraction data for COX enzyme complexes with NSAIDs (e.g., Flurbiprofen, Ibuprofen, Celecoxib).
- Multipole Refinement: Perform charge-density analysis based on multipolar refinement to evaluate intermolecular interactions.
- Energy Calculation: Apply UBDB+EPMM to compute electrostatic interaction energies between the drug and specific amino acid residues (e.g., Arg120, Tyr355) in the enzyme active site.
- Selectivity Analysis: Compare energy profiles between COX-1 and COX-2 isoforms to elucidate structural determinants of selectivity.
- Key Outcomes: Reveals that while electrostatic interactions are fundamental, the complex interplay of hydrophobic effect and van der Waals forces also critically contributes to binding affinity and selectivity [10].
Schematic Workflows and Signaling Pathways
The following diagrams illustrate key experimental and conceptual frameworks for characterizing interactions on sensor surfaces.
Electrostatics-Driven Screening Workflow
Sensor-Analyte Interaction Mechanism
The objective comparison of electrostatic, hydrophobic, and van der Waals interactions reveals that each mechanism contributes uniquely to the performance of NSAID characterization methods. Electrostatic interactions, quantified through advanced computational and crystallographic methods, provide the highest fidelity for molecular discrimination and understanding COX-2 selectivity. Hydrophobic forces, critically linked to lipophilicity, are paramount for binding affinity in both biological targets and sensor surfaces. Van der Waals interactions, while weaker individually, collectively provide essential structural complementarity and binding stability. The most effective sensor platforms and characterization strategies synergistically integrate all three mechanisms. The experimental data and protocols presented provide a robust toolkit for researchers to quantify these interactions and guide the rational design of next-generation sensors with enhanced sensitivity and selectivity for NSAIDs.
Non-specific adsorption (NSA) represents a fundamental challenge in biosensor development, directly compromising analytical performance through false positives, signal drift, and reduced sensitivity. NSA occurs when non-target molecules—such as proteins, lipids, or cellular debris—accumulate on biosensing interfaces through physisorption mediated by hydrophobic interactions, electrostatic forces, hydrogen bonding, or van der Waals interactions [1] [18]. In complex biological samples like blood, serum, or sweat, this fouling phenomenon becomes particularly problematic, leading to inaccurate readings and unreliable data across diverse biosensing platforms [1] [19].
The persistence of NSA presents a significant barrier to the widespread adoption of biosensors in critical applications including clinical diagnostics, drug development, and environmental monitoring. Understanding the specific mechanisms through which NSA degrades sensor performance is essential for developing effective characterization methods and antifouling strategies. This guide systematically examines the consequences of NSA, compares its impact across different sensor platforms, and provides detailed experimental protocols for its quantification and mitigation, framed within contemporary research on surface characterization methodologies [18].
Core Mechanisms: How NSA Compromises Biosensor Performance
False Positives and Altered Background Signals
Non-specifically adsorbed molecules generate background signals that are often indistinguishable from specific binding events, leading to false positive results. In surface-based affinity biosensors like SPR and cantilever systems, NSA contributes directly to the measured signal amplitude, creating a positive baseline shift that erroneously suggests target analyte presence [1] [20]. For electrochemical aptamer-based (E-AB) biosensors, fouling molecules can restrict the conformational freedom of structure-switching aptamers, limiting their ability to undergo the structural changes required for target binding and signal generation [1].
Table 1: Types and Impacts of False Positives in Biosensors
| NSA Type | Mechanism | Impact on Signal | Common Sensor Platforms Affected |
|---|---|---|---|
| Vacant Space Adsorption | Molecules adsorb on unfunctionalized areas of sensor surface | Increased baseline signal, reduced dynamic range | SPR, Microfluidic, Electrochemical |
| Non-Immunological Site Binding | Adsorption to non-recognition regions of bioreceptors | Steric hindrance, altered binding kinetics | Immunosensors, Aptasensors |
| Accessible Immunological Site Binding | Adsorption to immunological sites without blocking antigen access | Partial signal inhibition, altered dose-response | ELISA, SPR Immunosensors |
| Blocking Immunological Site Binding | Adsorption to immunological sites that blocks antigen access | False negatives, reduced sensitivity | All affinity-based biosensors |
Signal Drift and Measurement Instability
Signal drift manifests as a time-dependent change in the biosensor output unrelated to analyte concentration. NSA progressively degrades the sensing interface through the accumulation of foulants, leading to continuous signal variation that complicates data interpretation [1] [19]. In electrochemical systems, fouling dramatically affects interfacial characteristics and electron transfer kinetics, while in optical sensors like SPR, adsorbed molecules alter the refractive index at the sensing surface [1]. Over extended measurement periods, this drift can no longer be adequately corrected through background subtraction algorithms, fundamentally limiting sensor reliability for continuous monitoring applications [20].
Reduced Sensitivity and Dynamic Range Compression
NSA diminishes biosensor sensitivity through multiple mechanisms. Fouling molecules can physically block access to recognition elements, reduce mass transport to the sensing interface, and introduce additional resistance to electron transfer in electrochemical systems [1] [18]. This sensitivity loss manifests as a decreased slope in the dose-response curve and elevation of the limit of detection. In severe cases, NSA can completely passivate the sensing interface, leading to false negative results at low analyte concentrations [1]. The dynamic range also becomes compressed as the upper detection limit is reduced due to limited available binding sites, while the lower limit increases due to elevated background signals [20].
Quantitative Comparison: NSA Impacts Across Biosensor Platforms
Table 2: Comparative Impact of NSA on Major Biosensor Platforms
| Sensor Platform | Primary NSA Impact | Signal Change Mechanism | Typical Performance Degradation | Characterization Methods |
|---|---|---|---|---|
| Electrochemical (EC) | Altered electron transfer kinetics, surface passivation | Current/voltage modulation, increased charge transfer resistance | 50-400% signal suppression in sweat samples [19] | EIS, CV, Amperometry/Potentiometry |
| Surface Plasmon Resonance (SPR) | Refractive index change from adsorbed mass | Resonance angle/position shift | Indistinguishable from specific binding signals [1] | Resonance monitoring, reference channel subtraction |
| Piezoelectric Cantilever | Mass loading on resonant structure | Resonant frequency shift (Δf) | False classification rates up to 22% without ML correction [20] | Frequency spectrum analysis, Q-factor measurement |
| Electrochemical-SPR (EC-SPR) | Combined EC and optical effects | Simultaneous current and angle changes | Complex interference patterns requiring multivariate analysis [1] | Synchronized EC-SPR measurement |
| Wearable Sweat Sensors | Biofouling from complex matrix | Current drift, sensitivity loss | >50% signal suppression in real-use conditions [19] | Continuous monitoring with control sensors |
Advanced Characterization and Mitigation Methodologies
Experimental Protocols for NSA Quantification
Protocol 1: Dynamic Response Analysis with Machine Learning Classification
This protocol enables NSA quantification through classification of time-series biosensor data [20]:
Biosensor Functionalization: Immobilize appropriate bioreceptors (antibodies, aptamers, enzymes) following standard protocols for the specific sensor platform.
Data Acquisition: Expose sensors to calibration standards and complex samples (serum, sweat, milk) under continuous flow conditions. Record dynamic response (e.g., resonant frequency for cantilevers, current for electrochemical sensors, angle shift for SPR) with high temporal resolution.
Signal Normalization: Process raw signals using the equation: θ(t) = (S(t) - Si)/(Sf - Si), where S(t) is the instantaneous signal, Si is the initial baseline, and S_f is the final steady-state signal.
Feature Engineering: Extract both theory-guided features (binding rate constants, initial slope, time constants) and TSFRESH-based features (statistical, temporal characteristics) from the normalized dynamic response.
Machine Learning Classification: Implement supervised learning models (Random Forest, SVM) with stratified k-fold cross-validation (k=5) to classify responses based on analyte concentration and identify false positives/negatives resulting from NSA.
Protocol 2: Antifouling Hydrogel Coating Evaluation for Wearable Sensors
This protocol characterizes NSA resistance of novel coating materials [19]:
Material Synthesis: Prepare peptide composite hydrogels incorporating catalytic nanomaterials (e.g., Au-PdNPs/rGO) and engineered hydrophilic peptides at optimal concentration (6.5 mg·mL⁻¹).
Sensor Fabrication: Deposit hydrogel onto electrode surfaces using controlled deposition techniques (spin-coating, drop-casting) with defined thickness.
Surface Characterization: Quantify hydrophilicity via water contact angle measurement (target: <10°), and characterize surface morphology by SEM/AFM.
Antifouling Assessment: Expose coated sensors to undiluted human sweat or 10% serum solution for extended periods (2-24 hours) under static and flow conditions.
Performance Metrics: Calculate signal loss percentage = [(Iinitial - Ifinal)/I_initial] × 100, with <10% loss indicating excellent antifouling performance. Compare sensitivity retention before and after fouling challenges.
Multimodal Sensing for NSA Discrimination
Advanced biosensing platforms now employ multimodal detection to discriminate specific signals from NSA contributions. Integrated electrochemical/colorimetric/photothermal systems enable cross-validation through independent signal channels [21]. When one modality is compromised by fouling, the remaining channels maintain functionality, significantly reducing false positives. This approach leverages multifunctional nanozymes with complementary catalytic and photothermal properties, creating robust sensing systems that can identify NSA through discordance between different signal types [21].
Table 3: Research Reagent Solutions for NSA Characterization and Mitigation
| Reagent/Material | Composition | Function in NSA Research | Application Examples |
|---|---|---|---|
| Peptide Composite Hydrogel | Engineered hydrophilic peptides, Au-PdNPs/rGO nanohybrids | Antifouling coating with extreme hydrophilicity (9.01° contact angle) | Wearable sweat sensors (8.3% signal loss in undiluted sweat) [19] |
| Bovine Serum Albumin (BSA) | Serum albumin protein | Passive blocking agent for unmodified surfaces | Reduction of vacant space adsorption in ELISA, Western blot [18] |
| Theory-Guided Feature Set | 14 mathematically derived parameters from binding kinetics | Machine learning input for NSA identification | Classification of dynamic biosensor response with enhanced accuracy [20] |
| Multifunctional Nanozymes | Co/La MOF@Au, Au-Mn₃O₄ nanocomposites | Tri-modal signal generation (EC/colorimetric/photothermal) | Cross-validation and false-positive reduction in complex samples [21] |
| Boron-Doped Graphdiyne (BGDY) | Two-dimensional carbon nanomaterial with boron heteroatoms | Stable electrode substrate with enhanced conductivity | Improved signal stability in enzymatic biofuel cells [21] |
The consequences of NSA—false positives, signal drift, and reduced sensitivity—represent interconnected challenges that require multifaceted solutions. Effective management strategies now integrate advanced materials science with computational approaches, combining novel antifouling coatings with machine learning-enabled signal processing [20] [19]. The development of standardized characterization protocols and multimodal sensing platforms provides researchers with powerful tools to quantify and mitigate NSA impacts, advancing the reliability of biosensors across drug development, clinical diagnostics, and environmental monitoring applications. As these technologies mature, the integration of real-time NSA correction and self-validating sensing systems will further enhance biosensor performance in complex biological environments.
The performance of biosensors is critically dependent on the complex interplay between the sensor surface and the sample matrix in which measurements occur. Non-specific adsorption represents a significant challenge, leading to elevated background signals, reduced sensitivity, and false-positive results [18]. This phenomenon occurs when biomolecules physisorb to sensing surfaces through intermolecular forces such as hydrophobic interactions, ionic bonds, van der Waals forces, and hydrogen bonding [18]. The composition of the sample matrix—whether blood, serum, or environmental water—profoundly influences the extent and impact of NSA due to fundamental differences in complexity, protein content, and interfering substances. Understanding these matrix-specific effects is essential for developing robust sensing platforms for applications ranging from clinical diagnostics to environmental monitoring.
Sample Matrix Composition and NSA Challenges
The sample matrix introduces unique challenges for NSA reduction in biosensing applications. Blood, serum, and environmental water differ dramatically in their composition, each presenting distinct interferents that can compromise sensor performance.
- Blood: As a whole biological fluid, blood contains cellular components (red blood cells, white blood cells, platelets) suspended in plasma, along with a high concentration of proteins (approximately 60-80 g/L), lipids, electrolytes, and various metabolites [18]. This complexity makes blood particularly prone to NSA, as numerous components can physisorb to sensor surfaces.
- Serum: Serum represents the acellular fraction of blood after coagulation, lacking fibrinogen but retaining most other proteins, including albumin, immunoglobulins, and complement proteins [18]. With protein concentrations typically ranging from 60-80 g/L, serum remains a challenging matrix for biosensing despite the removal of cellular components.
- Environmental Water: This matrix encompasses diverse sources from drinking water to surface waters, containing variable levels of dissolved organic matter, inorganic ions, microorganisms, and anthropogenic contaminants such as per- and polyfluoroalkyl substances (PFAS) [22] [23]. While typically having lower overall complexity than biological fluids, environmental water can still present significant NSA challenges due to the presence of natural organic matter and other interferents.
Table 1: Key Characteristics of Different Sample Matrices Relevant to NSA
| Matrix | Primary Components | Typical Protein Content | Major NSA Contributors |
|---|---|---|---|
| Blood | Cells, platelets, plasma proteins, lipids, electrolytes | 60-80 g/L | Cellular components, albumin, immunoglobulins, fibrinogen |
| Serum | Albumin, globulins, electrolytes, hormones, metabolites | 60-80 g/L | Albumin, immunoglobulins, complement proteins |
| Environmental Water | Dissolved organic matter, inorganic ions, microorganisms, contaminants | Negligible | Natural organic matter, humic acids, PFAS, microbial content |
Comparative Analysis of NSA Across Matrices
Quantitative NSA Assessment in Environmental Water Monitoring
Studies measuring PFAS in environmental water demonstrate the critical importance of matrix considerations in analytical sensitivity. Research on PFAS contamination in the Haw River, North Carolina, revealed extensive contamination with both legacy and emerging PFAS compounds [22]. The study implemented rigorous protocols to minimize NSA and interference during sample collection and analysis. Pre-cleaned high-density polyethylene bottles were prepared with sequential rinses of methanol, ammonium hydroxide, and ultra-pure water to reduce background contamination [22]. For LC-MS/MS analysis, solid-phase extraction concentrated PFAS from 800 mL water samples while reducing matrix effects through selective binding and washing steps [22].
Table 2: PFAS Detection in Paired Water and Serum Samples from Pittsboro, NC
| PFAS Compound | Detection in Drinking Water | Detection in Serum | Notable Matrix Effects |
|---|---|---|---|
| PFHxA | Highest concentrations measured | Detected, reflecting temporal variability in water | Shorter half-life demonstrates matrix-specific accumulation differences |
| PFOA | Detected (historical levels up to 287 ng/L) | Detected in all participants, 2-4x U.S. median | Serum levels reflect historical exposure due to long half-life |
| PFOS | Detected (historical levels up to 132 ng/L) | Detected in all participants, 2-4x U.S. median | Current water levels not associated with current serum levels |
| PFHxS | Detected | Detected in all participants | Associated with increased total and non-HDL cholesterol |
Serum Matrix Considerations in Clinical Exposure Assessment
The California Regional Exposure study highlighted the relationship between PFAS in public water systems and serum concentrations in a general population [23]. This research demonstrated that even at lower exposure levels, PFAS detections in drinking water were associated with higher serum concentrations, with PFHxS geometric mean concentrations 31.9% higher among participants with detectable PFHxS in their water [23]. The study employed sophisticated serum processing methods to minimize NSA, including online solid-phase extraction coupled with ultra-high performance liquid chromatography tandem mass spectrometry (SPE-HPLC-MS/MS) [23]. Serum samples were processed using C18 extraction cartridges followed by separation on C8 HPLC columns before MRM analysis, effectively reducing matrix interference [23].
Methodologies for NSA Reduction Across Matrices
Passive NSA Reduction Methods
Passive methods aim to prevent undesired adsorption by coating the sensor surface with physical or chemical barriers [18]. These approaches have been extensively developed over decades and represent the first line of defense against NSA.
- Physical Blocking: The most common physical method uses blocker proteins such as bovine serum albumin (BSA), casein, and other milk proteins that adsorb to surfaces, creating a protective layer that reduces non-specific binding [18]. These are particularly effective for serum and blood matrices where they compete with endogenous proteins for binding sites.
- Chemical Surface Modification: Chemical methods employ linker molecules and polymers to create thin, hydrophilic, and non-charged boundary layers that thwart protein adsorption [18]. Materials for these non-fouling coatings are typically neutral or weakly negative and well-hydrated, creating a thermodynamic barrier to adsorption.
Active NSA Reduction Methods
Active methods dynamically remove adsorbed molecules after functionalization and represent a more recent technological advancement, particularly valuable for complex matrices like blood and serum [18].
- Transducer-Based Removal: These methods use electromechanical or acoustic transducers to generate surface forces that shear away weakly adhered biomolecules [18]. The mechanical energy disrupts the physisorption bonds responsible for NSA.
- Hydrodynamic Removal: This approach relies on pressure-driven flow in microfluidic systems to create shear forces that remove non-specifically bound molecules [18]. The method is particularly compatible with environmental water samples where volume constraints are less limiting.
Experimental Protocols for NSA Characterization
Serum Sample Processing and PFAS Analysis Protocol
The CARE study implemented a detailed protocol for serum PFAS analysis that effectively minimizes matrix-derived NSA [23]:
- Sample Collection: Licensed phlebotomists collected blood samples, which were centrifuged to separate serum. Serum samples were stored at -20°C and shipped on dry ice to prevent degradation.
- Sample Extraction: Serum samples were processed using an online Symbiosis Pharma SPE-HPLC system with Mistral CS Cool. Samples were loaded onto HySphere C18 HD cartridges (10 × 2 mm, 7 μm).
- Wash and Elution: After loading, cartridges were washed to remove interfering matrix components, then target analytes were eluted onto a BETASIL C8 HPLC column for separation.
- Detection and Quantification: The eluate was introduced to a Sciex 4000 QTrap mass spectrometer operating in multiple-reaction-monitoring mode. Quantification used the area of Q1/Q3 ion pairs with isotope dilution for precision.
- Quality Assurance: Method accuracy was verified using NIST Standard Reference Material 1958, with duplicate quality control samples in each batch and regular participation in external proficiency testing.
Biosensor Functionalization and Testing Protocol
Advanced Silicon Group developed a biosensor protocol that addresses matrix challenges through silicon nanowire technology [24]:
- Sensor Fabrication: Silicon nanowires are fabricated on chips, creating structures sensitive to surface charge changes.
- Surface Functionalization: Nanowires are functionalized with antibodies specific to target proteins through silane chemistry, creating a capture surface.
- Sample Application: A small volume of sample (serum, blood, or environmental water) is applied to the sensor surface.
- Incubation and Rinsing: The sensor is incubated to allow specific binding, followed by rinsing to remove non-specifically adsorbed components.
- Detection: The functionalized silicon nanowire sensor detects bound proteins through changes in photocurrent when exposed to light, as bound proteins alter carrier recombination in the silicon.
- Quantification: The photocurrent change is correlated with protein concentration, providing quantification within 15 minutes with significantly reduced cost compared to ELISA [24].
Diagram 1: Experimental Framework for Matrix-Specific NSA Characterization. This workflow illustrates the relationship between sample matrices, NSA reduction methods, and detection technologies.
Research Reagent Solutions for NSA Characterization
Table 3: Essential Research Reagents for NSA Reduction in Different Matrices
| Reagent/Material | Function | Application Specifics |
|---|---|---|
| Bovine Serum Albumin (BSA) | Physical blocking agent that adsorbs to surfaces, reducing non-specific protein binding | Effective for blood and serum matrices; typically used at 1-5% concentration in incubation buffers |
| Casein | Protein-based blocking agent derived from milk, effective at reducing NSA in immunoassays | Particularly useful for serum applications; forms a protective layer on sensor surfaces |
| C18 Extraction Cartridges | Solid-phase extraction medium for concentrating analytes and removing matrix interferents | Critical for environmental water PFAS analysis; used in 10 × 2 mm, 7 μm configurations |
| Silicon Nanowire Sensors | Functionalized biosensors with antibodies for specific protein detection while minimizing NSA | Enable rapid (15-minute) testing with reduced cost; compatible with multiple matrices |
| HySphere C18 HD | Solid-phase extraction material with high retention capacity for PFAS compounds | Used in 7 μm particle size, 10 × 2 mm dimensions for serum PFAS extraction |
| Self-Assembled Monolayers (SAMs) | Chemical surface modification that creates ordered molecular films to resist NSA | Form well-defined interfaces that reduce physisorption in complex matrices |
| BETASIL C8 Columns | HPLC separation columns providing optimal resolution of PFAS compounds from matrix components | Used for final separation before MS/MS detection in serum analysis |
The critical role of the sample matrix in biosensing applications cannot be overstated, with blood, serum, and environmental water each presenting distinct challenges for NSA reduction. Effective characterization and mitigation of NSA require matrix-specific strategies that account for fundamental differences in composition and interferents. Passive methods using blocker proteins and chemical surface modifications provide essential foundational protection, while active removal methods offer dynamic NSA reduction for particularly challenging matrices. The continuing advancement of biosensor technologies, including silicon nanowire platforms and improved chromatographic methods, enables more effective NSA management across diverse sample types. As regulatory standards for contaminants like PFAS become increasingly stringent [23], the development of matrix-optimized NSA reduction strategies will remain essential for accurate exposure assessment and health effects research.
Analytical Techniques for NSA Quantification: From Electrochemistry to Plasmonics
The performance of an electrochemical sensor is fundamentally governed by the processes occurring at the interface between the electrode surface and the analyte solution. For research focused on characterizing methods for quantifying non-specific adsorption (NSA) on sensor surfaces, understanding and controlling this interface is paramount. Non-specific adsorption can severely compromise sensor selectivity and lead to false positives, making its accurate quantification essential for developing reliable diagnostic tools. Electrochemical methods provide a powerful, label-free means to probe this interface in real-time, offering insights into the kinetics, thermodynamics, and integrity of the surface layer. Techniques such as Electrochemical Impedance Spectroscopy (EIS), Cyclic Voltammetry (CV), and Differential Pulse Voltammetry (DPV) are cornerstone methods for this characterization, each providing complementary information on surface properties, binding events, and the extent of non-specific fouling.
The selection of an appropriate electrochemical technique is dictated by the specific parameter of interest—be it the capacitive nature of an antifouling layer, the electron transfer kinetics of a redox probe, or the sensitive quantification of an adsorbed target molecule. This guide provides a comparative analysis of EIS, CV, and DPV, equipping researchers and drug development professionals with the knowledge to select the optimal method for quantifying NSA and advancing sensor surface research.
Core Principles of Key Electrochemical Techniques
Electrochemical techniques function by applying an electrical signal to an electrochemical cell and measuring the resulting response. The three techniques discussed here—EIS, CV, and DPV—differ in the nature of the input signal and the resulting output, which in turn dictates the type of information that can be extracted about the electrode-solution interface.
Cyclic Voltammetry (CV) is a potentiodynamic technique where the potential applied to the working electrode is scanned linearly between two set limits and then scanned back. The resulting current is plotted against the applied potential to produce a voltammogram. This method is highly effective for studying the redox behavior of electroactive species, assessing the reversibility of reactions, and characterizing modified electrode surfaces. In CV, the continuous potential scan provides information on both oxidation and reduction processes in a single cycle, with key parameters including peak potentials (Epa, Epc) and peak currents (Ipa, Ipc) [25] [26]. The shape of the voltammogram can reveal the kinetics of electron transfer; a reversible system shows a characteristic pair of peaks, while a suppressed current often indicates a passivated or blocked surface [25].
Differential Pulse Voltammetry (DPV), another potentiodynamic technique, is designed to enhance sensitivity and lower detection limits. In DPV, small amplitude potential pulses are superimposed on a linear potential ramp. The current is sampled twice for each pulse—just before the pulse is applied and at the end of the pulse. The difference between these two current measurements is plotted against the base potential. This differential current measurement effectively minimizes the contribution of the capacitive (charging) current, which is non-faradaic, thereby amplifying the faradaic current related to the redox reaction of the analyte. This makes DPV exceptionally well-suited for the trace-level detection of analytes and for quantifying surface-bound species, such as in molecularly imprinted polymer (MIP) sensors [27] [25].
Electrochemical Impedance Spectroscopy (EIS) operates in the frequency domain rather than the time or potential domain. It measures the impedance (Z), or opposition to current flow, of an electrochemical system over a wide range of frequencies in response to a small amplitude alternating current (AC) potential. The resulting data is often presented in a Nyquist plot and interpreted using equivalent electrical circuit models. EIS is unparalleled for probing the dielectric and resistive properties of the electrode interface. It is particularly powerful for characterizing the formation of insulating layers on electrodes, such as self-assembled monolayers or polymer films, and for monitoring binding events that alter the charge transfer resistance (Rct), such as the adsorption of proteins or other biomolecules. This makes it an indispensable tool for studying non-specific adsorption and the integrity of antifouling coatings [28] [26].
Table 1: Core Operational Principles of EIS, CV, and DPV.
| Technique | Input Signal | Measured Output | Primary Data Format |
|---|---|---|---|
| EIS | Small AC potential over a frequency range | Impedance (Z) and Phase Angle (θ) | Nyquist Plot, Bode Plot |
| CV | Linear potential sweep reversed at vertex | Current (I) | Voltammogram (I vs. E) |
| DPV | Linear potential ramp with superimposed pulses | Differential Current (ΔI) | Peak Plot (ΔI vs. E) |
Comparative Analysis of Technique Performance
The utility of EIS, CV, and DPV becomes evident when their analytical performance is compared directly. Each technique excels in different aspects, making them suitable for specific stages of sensor characterization and application.
Sensitivity and Limit of Detection (LOD): DPV is renowned for its high sensitivity and low LOD. By minimizing the capacitive current, it can resolve very small faradaic currents from trace analytes. For instance, in the detection of glycerol using a molecularly imprinted polymer sensor, a remarkably low LOD of 0.001 μg/mL was achieved using DPV, which was significantly better than what was attainable with EIS on the same sensor [27]. CV, while excellent for qualitative studies, is generally less sensitive than DPV due to the higher background capacitive current. EIS does not provide a direct "detection limit" in the same way; its sensitivity is reflected in the ability to measure minute changes in interfacial properties, such as a small increase in charge transfer resistance (Rct) due to a binding event.
Information Depth and Interface Characterization: EIS provides the most comprehensive picture of the electrochemical interface. It can deconvolute different physical processes occurring at various frequencies. High-frequency data often relates to solution resistance and geometric capacitance, while low-frequency data probes diffusion and charge transfer kinetics. This allows researchers to build an equivalent circuit model that describes the interface in detail, including solution resistance (Rs), charge transfer resistance (Rct), and double-layer capacitance (Cdl) [26]. In contrast, CV and DPV primarily provide information about faradaic processes and the effective surface area.
Speed and Throughput: CV and DPV are generally faster for a single measurement, with a typical CV scan taking minutes. DPV scans can be slower than CV due to the pulse sequence but require fewer scans for quantification. EIS is the most time-consuming technique, as it requires measurements across a wide range of frequencies, which can take from several minutes to hours depending on the low-frequency limit and the system's stability.
Table 2: Analytical Performance Comparison for Interface Characterization.
| Parameter | EIS | CV | DPV |
|---|---|---|---|
| Limit of Detection | Moderate (indirect) | Moderate | Very Low [27] [25] |
| Quantification of NSA | Excellent (via Rct/Cdl changes) | Good (via current suppression) | Good (via peak diminishment) |
| Kinetic Information | Electron transfer kinetics, diffusion | Electron transfer kinetics, reaction reversibility | Limited kinetic information |
| Measurement Speed | Slow | Fast | Moderate |
| Ease of Data Interpretation | Complex (requires modeling) | Moderate (direct visual cues) | Simple (direct peak analysis) |
Experimental Protocols for Method Validation
To ensure reliable and reproducible data for quantifying NSA, standardized experimental protocols are critical. The following sections outline general methodologies for employing EIS, CV, and DPV in sensor surface characterization.
Protocol for Baseline Characterization with CV
This protocol establishes a baseline for electrode surface area and cleanliness, which is a prerequisite for NSA studies.
- Electrode Preparation: Polish the working electrode (e.g., glassy carbon) with successively finer alumina slurries (e.g., 1.0, 0.3, and 0.05 μm) on a microcloth pad. Ruminate thoroughly with deionized water between each polish and sonicate for 1-2 minutes to remove adsorbed alumina particles.
- Redox Probe Preparation: Prepare a solution of 5 mM Potassium Ferricyanide (K₃[Fe(CN)₆]) in 1 M Potassium Chloride (KCl) supporting electrolyte. The KCl ensures high ionic strength to minimize solution resistance.
- Data Acquisition: Fill the electrochemical cell with the redox probe solution and deaerate with an inert gas (e.g., N₂ or Ar) for 10-15 minutes. Assemble the three-electrode system (working, reference, counter). Run CV scans, for example, between -0.1 V and +0.5 V vs. Ag/AgCl, at a scan rate of 50 mV/s until stable, reproducible voltammograms are obtained. A well-defined, symmetric redox peak with a small peak separation (ΔEp ≈ 59/n mV) indicates a clean, electrochemically reversible surface [25] [29].
- Data Analysis: Calculate the electroactive surface area using the Randles-Ševčík equation: Ip = (2.69×10⁵) * n^(3/2) * A * D^(1/2) * C * v^(1/2), where Ip is the peak current, n is electrons transferred, A is the area (cm²), D is the diffusion coefficient, C is the concentration (mol/cm³), and v is the scan rate (V/s).
Protocol for Surface Modification Monitoring with EIS
EIS is ideal for monitoring the step-by-step build-up of a sensor surface and the subsequent NSA.
- Initial EIS Measurement: Using the same [Fe(CN)₆]³⁻/⁴⁻ redox probe, perform an EIS measurement on the clean, characterized electrode. A typical setup applies a DC potential equivalent to the formal potential of the redox couple (often ~0.22 V vs. Ag/AgCl) with an AC potential amplitude of 5-10 mV, sweeping frequencies from 100 kHz to 0.1 Hz.
- Surface Modification: Incubate the electrode in the solution containing the surface modifier (e.g., a thiol-based self-assembled monolayer, a polymer, or a recognition element) for a specified time.
- Post-Modification EIS Measurement: After thoroughly rinsing the electrode, record a new EIS spectrum in the fresh redox probe solution.
- NSA Challenge and Quantification: Incubate the modified electrode in a complex matrix (e.g., serum, blood, or a solution of a non-target protein like BSA) to induce non-specific adsorption. Rinse and perform a final EIS measurement.
- Data Analysis: Fit all EIS spectra to a suitable equivalent circuit, such as R(QR)(QR) or a modified Randles circuit. The key parameter to monitor is the charge transfer resistance (Rct). A successful modification that passivates the surface will cause a large increase in Rct. A further increase after the NSA challenge quantitatively indicates the level of fouling, as adsorbed species further hinder the redox probe's access to the electrode. Changes in the constant phase element (CPE) can also provide information on the capacitive nature of the interface.
Protocol for Sensitive Quantification with DPV
DPV is used for highly sensitive measurement of surface-bound or solution-phase analytes after surface modification.
- Parameter Setup: Configure the DPV parameters on the potentiostat. Typical settings include a pulse amplitude of 25-50 mV, a pulse width of 50 ms, and a step potential of 1-10 mV.
- Calibration: For quantitative analysis, record DPV scans in standard solutions of the target analyte with known concentrations. A background scan in pure supporting electrolyte may be subtracted.
- Sample Measurement: After exposing the sensor to the sample solution (or after an NSA challenge if the fouling agent is electroactive), rinse the electrode and transfer it to a clean electrochemical cell containing a supporting electrolyte. Run the DPV scan.
- Data Analysis: The faradaic current will manifest as a peak. Plot the peak height (ΔI) against the analyte concentration for the calibration curve. The concentration of an unknown sample or the extent of adsorption can be determined by interpolating from this curve [27].
Visualization of Experimental Workflows
The following diagrams illustrate the logical workflow for sensor characterization and the fundamental signal structures of each electrochemical technique.
The Scientist's Toolkit: Essential Research Reagents and Materials
The following table details key reagents, materials, and instruments essential for conducting rigorous electrochemical characterization of sensor surfaces.
Table 3: Essential Research Reagents and Materials for Electrochemical Characterization.
| Item Name | Function/Application | Key Characteristics |
|---|---|---|
| Potentiostat/Galvanostat | Core instrument for applying potentials/currents and measuring electrochemical signals. | Precision, stability, modularity (e.g., AMEL, PalmSens) [26]. |
| Screen-Printed Electrodes (SPEs) | Disposable, integrated three-electrode cells for rapid testing and portability. | Low-cost, mass-producible, various substrate materials (ceramic, paper) [29]. |
| Potassium Ferricyanide/Ferrocyanide | Standard redox probe for characterizing electrode surface area and electron transfer kinetics. | Reversible, well-understood electrochemistry [25] [29]. |
| Gold Nanoparticles (AuNPs) | Nanomaterial for electrode modification to enhance conductivity and active surface area. | High conductivity, biocompatibility, facile functionalization [27]. |
| Molecularly Imprinted Polymers (MIPs) | Synthetic receptors for creating highly selective recognition sites on sensor surfaces. | High specificity, stability, cost-effectiveness compared to biological receptors [27]. |
| Phosphate Buffered Saline (PBS) | Common supporting electrolyte for biochemical sensing; provides stable pH and ionic strength. | Physiological pH (7.4), non-corrosive, biocompatible. |
Surface Plasmon Resonance (SPR) and Localized Surface Plasmon Resonance (LSPR) represent two cornerstone techniques in the realm of real-time, label-free biomolecular interaction analysis. These optical sensing platforms have revolutionized how researchers monitor binding events on sensor surfaces, providing invaluable insights into interaction kinetics, affinity, and specificity. SPR technology exploits the collective oscillation of free electrons at the interface between a metal (typically gold) and a dielectric medium, which generates surface plasmon waves that are exquisitely sensitive to changes in the local refractive index [30] [31]. When biomolecular binding occurs on the sensor surface, it alters the refractive index, causing a detectable shift in the resonance conditions that can be monitored in real-time without requiring fluorescent or radioactive labels [31] [32].
LSPR operates on a related but distinct principle, where the resonant oscillations are confined to metallic nanostructures rather than propagating along a continuous metal film [30]. This fundamental difference in physical mechanism translates to significant practical implications for their application in binding monitoring. SPR typically offers higher sensitivity to bulk refractive index changes and provides detailed kinetic information, while LSPR exhibits superior spatial resolution and is more tolerant to temperature fluctuations [30]. Both techniques have found extensive application across diverse fields including pharmaceutical development, clinical diagnostics, environmental monitoring, and fundamental biological research, enabling the characterization of interactions between various biomolecules such as antibodies and antigens, DNA and proteins, and small molecule drugs and their targets [30] [31] [32]. The evolution of these technologies continues to advance with recent developments in nanotechnology and microfluidics, further enhancing their capabilities and accessibility [33].
Fundamental Principles and Theoretical Background
Surface Plasmon Resonance (SPR) Physics
The physical foundation of SPR technology centers on the phenomenon of surface plasmons – coherent electron oscillations that propagate along the interface between a metal and a dielectric material. In practical sensing applications, SPR is most commonly excited using the Kretschmann configuration, where light is directed through a prism onto a thin metal film (typically gold) under conditions of total internal reflection [34] [31]. At a specific angle of incidence known as the resonance angle, the momentum of the incident photons couples with the surface plasmons, resulting in a transfer of energy that manifests as a sharp dip in the intensity of reflected light [31]. This resonance condition is highly sensitive to changes in the refractive index within the evanescent field, which typically extends 100-300 nanometers from the metal surface [34].
The underlying physics can be mathematically described by several key equations. The propagation constant of surface plasmons is given by: ( k{SPP} = k0 \sqrt{\frac{\varepsilonm \varepsilond}{\varepsilonm + \varepsilond}} ) where ( k0 ) is the wave vector of incident light, ( \varepsilonm ) is the dielectric constant of the metal, and ( \varepsilond ) is the dielectric constant of the dielectric medium [34]. The resonance condition is achieved when the wave vector component of the incident light parallel to the interface matches this surface plasmon propagation constant. In sensing applications, when biomolecules bind to the functionalized metal surface, they alter the local refractive index (( \varepsilond )), leading to a measurable shift in the resonance angle that is directly proportional to the mass concentration of bound analyte [31] [32]. This relationship forms the quantitative basis for SPR biosensing, enabling real-time monitoring of binding interactions with exceptional sensitivity.
Localized Surface Plasmon Resonance (LSPR) Fundamentals
In contrast to propagating surface plasmons, LSPR involves non-propagating plasmon oscillations that are confined to metallic nanoparticles or nanostructures with dimensions smaller than the wavelength of incident light [30]. When illuminated, the conduction electrons in these nanostructures collectively oscillate at a frequency resonant with the incident electromagnetic field, creating enhanced local fields near the particle surfaces. The LSPR extinction spectrum – characterized by a distinct absorption peak – is determined by multiple factors including the nanoparticle's composition, size, shape, and the local dielectric environment [30].
The resonance condition for LSPR can be described by the expression: ( \varepsilon'(\omega) = -2\varepsilonm ) where ( \varepsilon'(\omega) ) is the real part of the metal's frequency-dependent dielectric function and ( \varepsilonm ) is the dielectric constant of the surrounding medium [30]. This simplified expression applies to small, spherical nanoparticles in the quasi-static limit. The extreme sensitivity of LSPR to local environmental changes stems from the dependence of the resonance wavelength on ( \varepsilon_m ). When target analytes bind to functionalized nanoparticles, they alter the local dielectric environment, producing measurable spectral shifts in the LSPR peak position [30]. The magnitude of this shift depends on the size and conformation of bound molecules and their proximity to the nanoparticle surface, with the strongest effects occurring within the first approximately 10-30 nanometers. This distance dependence, coupled with the absence of propagating waves, differentiates LSPR from conventional SPR and defines its unique application profile in binding monitoring.
Table 1: Comparison of Fundamental Properties Between SPR and LSPR
| Property | SPR | LSPR |
|---|---|---|
| Plasmon Type | Propagating surface plasmons | Localized surface plasmons |
| Sensing Volume | ~100-300 nm evanescent field [34] | <30 nm from nanoparticle surface [30] |
| Measurement | Resonance angle shift [31] [32] | Extinction peak wavelength shift [30] |
| Setup | Prism-coupled (Kretschmann) [34] [31] | Direct illumination of nanostructures [30] |
| Temperature Sensitivity | High (requires precise temperature control) | Moderate (less susceptible to bulk effects) |
Technical Comparison of SPR and LSPR Platforms
Performance Metrics and Sensing Capabilities
The practical implementation of SPR and LSPR technologies reveals distinct performance characteristics that determine their suitability for specific applications. SPR systems typically exhibit exceptional sensitivity to bulk refractive index changes, with detection limits capable of reaching 10-12 mol/L for certain analytes [31]. This high sensitivity enables the detection of low molecular weight compounds and subtle binding interactions that would be challenging to monitor with other techniques. SPR's response is linear with mass accumulation on the sensor surface, making it particularly well-suited for quantitative analysis of binding affinities and kinetics [31] [32]. However, this bulk sensitivity also renders SPR measurements more vulnerable to temperature fluctuations and non-specific binding in complex media, often necessitating sophisticated temperature control and careful sample preparation.
LSPR platforms generally demonstrate slightly lower absolute sensitivity to bulk refractive index changes but offer enhanced sensitivity to local binding events occurring in immediate proximity to the nanoparticle surface [30]. This characteristic makes LSPR particularly valuable for detecting small molecules and monitoring conformational changes in bound proteins. The typical detection limit for LSPR sensors falls in the picomolar to nanomolar range for protein analytes, though this varies significantly with nanoparticle geometry and composition [30]. A key advantage of LSPR systems is their substantially reduced susceptibility to temperature drift, as the resonant excitation is less affected by bulk solvent effects compared to SPR. Additionally, LSPR platforms do not require the precise optical alignment and bulky coupling components of traditional SPR instruments, facilitating the development of compact, portable sensing devices suitable for point-of-care applications [33].
Instrumentation and Implementation
Traditional SPR instrumentation employs sophisticated optical systems including high-precision angle scanning mechanisms or imaging detectors to monitor resonance changes [34] [32]. Commercial platforms such as Biacore and Sierra SPR Pro systems incorporate microfluidics for automated sample delivery and temperature regulation to maintain measurement stability [31] [32]. These systems typically use sensor chips with a continuous ~50 nm gold film functionalized with various chemistries to immobilize specific binding partners [31]. The complexity of these optical and fluidic systems contributes significantly to the cost of traditional SPR instruments, which often range from $200,000 to $500,000, potentially limiting their accessibility for smaller laboratories [33].
LSPR instrumentation is generally more compact and simpler in design, as it typically measures extinction or scattering spectra from nanoparticle substrates without requiring precise angular alignment [30]. Recent advances have further simplified LSPR readout systems, with some platforms utilizing conventional microscopy equipment for signal detection [33]. The emergence of 3D nanoplasmonic structures has dramatically enhanced signal intensity in LSPR systems, enabling detection with basic optical components and significantly reducing costs [33]. These developments have facilitated the creation of portable LSPR-based sensors potentially costing less than $50,000, making the technology accessible to a broader user base [33]. Both platforms continue to evolve with integration of high-throughput capabilities, with modern systems supporting parallel analysis of multiple interactions through array-based formats and microfluidic automation [32].
Table 2: Comparison of Instrumentation and Practical Implementation
| Aspect | SPR | LSPR |
|---|---|---|
| Instrument Complexity | High (precision optics, microfluidics) [31] [32] | Moderate to low (simplified optics) [30] [33] |
| Cost | $200,000-$500,000 [33] | <$50,000 (newer systems) [33] |
| Throughput | Moderate (4-384 channels in advanced systems) [32] | High (array-based formats) [30] |
| Sample Consumption | Low (microfluidic delivery) [32] | Very low (small flow cells or static measurements) |
| Portability | Limited (benchtop systems) | High (compact systems possible) [33] |
Experimental Protocols and Methodologies
SPR Experimental Workflow for Binding Kinetics
A standardized SPR protocol for quantifying biomolecular interactions typically begins with sensor chip preparation, most commonly employing carboxymethylated dextran matrices on gold films for ligand immobilization [31]. The surface is activated using a mixture of N-ethyl-N'-(3-dimethylaminopropyl)carbodiimide (EDC) and N-hydroxysuccinimide (NHS) to create reactive esters for covalent coupling. The ligand (typically an antibody, antigen, or receptor protein) is then injected over the activated surface at concentrations ranging from 1-100 μg/mL in appropriate immobilization buffers, resulting in approximately 5-20 kDa of immobilized protein depending on the application requirements [31]. Remaining reactive groups are subsequently quenched with ethanolamine, and a series of conditioning pulses may be applied to stabilize the surface before analysis.
For binding kinetics measurements, the analyte is injected over the functionalized surface at a series of concentrations (typically spanning a 100-fold range) using continuous flow to maintain constant delivery [31] [32]. The association phase is monitored for 2-5 minutes, followed by dissociation monitoring in buffer flow for 5-60 minutes depending on the interaction stability. The resulting sensorgrams – plots of response units versus time – are processed by subtracting signals from reference flow cells to account for bulk refractive index changes and non-specific binding. The processed data is then fitted to appropriate binding models (e.g., 1:1 Langmuir, conformational change, or bivalent analyte models) using specialized software to extract kinetic parameters including the association rate constant (ka), dissociation rate constant (kd), and equilibrium dissociation constant (KD = kd/ka) [31]. Regeneration of the surface for subsequent analysis cycles is achieved using brief pulses of mild acidic or basic solutions (e.g., 10 mM glycine-HCl, pH 2.0-3.0) that disrupt the binding interaction without damaging the immobilized ligand.
Diagram 1: SPR Experimental Workflow for Binding Kinetics Analysis
LSPR Experimental Protocol for Binding Studies
LSPR binding studies employ nanostructured substrates rather than continuous metal films, with common configurations including colloidal nanoparticles in solution, nanoparticles immobilized on transparent substrates, or nanohole arrays in metal films [30]. Substrate functionalization typically begins with surface modification using self-assembled monolayers (SAMs) of alkanethiols on gold or silver nanoparticles, creating a bio-inert background while presenting specific functional groups (e.g., carboxyl, amine, or maleimide) for subsequent ligand attachment [30]. Ligand immobilization follows similar chemistry to SPR but often requires optimization of density to maintain nanoparticle stability and avoid aggregation. For quantitative binding studies, the LSPR extinction spectrum is measured before and after each functionalization step using ultraviolet-visible (UV-Vis) spectroscopy, with the peak wavelength position tracked precisely.
Binding experiments are performed by exposing the functionalized LSPR substrates to analyte solutions under controlled conditions, with spectral measurements taken at defined time intervals to monitor binding-induced wavelength shifts [30]. For kinetic analysis, continuous flow systems may be employed to maintain constant analyte concentration, though many LSPR platforms utilize static incubation due to their reduced sensitivity to bulk effects. Following data collection, the time-dependent wavelength shifts are converted to bound analyte concentrations using established calibration curves, enabling derivation of kinetic parameters similar to SPR [30]. The shorter sensing volume of LSPR (typically <30 nm from the nanoparticle surface) provides inherent filtering of bulk signal contributions, often eliminating the need for reference subtraction in well-designed experiments. Regeneration of LSPR sensors follows principles similar to SPR, though the chemical stability of the nanostructures must be considered when selecting regeneration conditions to prevent damage or alteration of the plasmonic properties.
Research Reagent Solutions and Materials
Successful implementation of SPR and LSPR binding studies requires careful selection of reagents and materials optimized for each platform. The core component for both technologies is the sensor surface, with SPR utilizing flat, ~50 nm gold films on glass substrates with refractive index typically around 1.61 [31] [32]. These surfaces are commonly modified with carboxymethylated dextran matrices that provide a hydrophilic, low-nonspecific binding environment while enabling covalent immobilization of ligands through amine, thiol, or other specific chemistries [31]. Specialized sensor chips are available for particular applications, including hydrophobic surfaces for membrane protein studies, nitrilotriacetic acid (NTA) surfaces for capturing histidine-tagged proteins, and streptavidin-coated surfaces for biotinylated ligands [32].
LSPR platforms employ diverse nanostructured materials ranging from spherical gold and silver nanoparticles to more complex geometries such as nanorods, nanostars, and nanocubes, each offering distinct plasmonic properties and tuning possibilities [30]. The resonance wavelength of these nanostructures depends critically on their composition, size, and shape, enabling customization for specific experimental needs. Surface functionalization typically uses alkanethiolate SAMs for gold-based nanostructures or silane chemistry for oxide-coated nanoparticles, with specific capture molecules then attached through similar chemistries as SPR [30]. Both technologies require high-quality running buffers with carefully controlled ionic strength and pH, often supplemented with low concentrations of surfactant (e.g., 0.005% Tween 20) to minimize non-specific binding without interfering with specific interactions [31]. Regeneration solutions vary depending on the interaction strength and stability of the immobilized ligand, with common choices including glycine-HCl (pH 2.0-3.0), NaOH (10-100 mM), and SDS (0.01-0.1%).
Table 3: Essential Research Reagents and Materials for SPR and LSPR
| Category | Specific Items | Function | SPR/LSPR Application |
|---|---|---|---|
| Sensor Surfaces | Gold film chips (~50 nm) [31] | Plasmon propagation substrate | SPR |
| Gold/silver nanoparticles (10-100 nm) [30] | Localized plasmon resonance | LSPR | |
| Immobilization Chemistry | Carboxymethylated dextran matrix [31] | Hydrophilic, low-nonspecific binding surface | SPR |
| Self-assembled monolayers (alkanethiols) [30] | Controlled surface functionalization | LSPR | |
| Coupling Reagents | EDC/NHS mixture [31] | Activates carboxyl groups for amine coupling | Both |
| Ethanolamine | Blocks remaining activated groups | Both | |
| Running Buffers | HBS-EP (10 mM HEPES, 150 mM NaCl, 3 mM EDTA, 0.005% P20) [31] | Standard running buffer with surfactant | Both |
| Regeneration Solutions | Glycine-HCl (10-100 mM, pH 2.0-3.0) [31] | Dissociates bound complexes without damaging ligand | Both |
Applications in Biomolecular Interaction Analysis
Pharmaceutical and Drug Development Applications
SPR and LSPR platforms have become indispensable tools in pharmaceutical research and development, particularly in the characterization of therapeutic candidates and lead optimization. SPR's ability to provide precise kinetic parameters (ka, kd, KD) makes it invaluable for ranking compound libraries during early-stage drug discovery [32]. The technology is extensively used for characterizing monoclonal antibodies, with applications ranging from epitope binning and affinity maturation studies to quality control of manufactured biologics [31] [32]. SPR can detect binding events involving diverse molecular classes including small molecules (<200 Da), peptides, proteins, nucleic acids, and even entire viruses or bacterial cells [32]. The real-time, label-free nature of SPR provides critical insights into binding mechanisms that might be obscured by labels in other techniques, such as conformational changes or complex multivalent interactions.
LSPR's unique attributes have enabled distinct applications in drug development, particularly in scenarios where small molecule detection or spatial resolution is paramount [30]. The technology's compact format facilitates high-throughput screening campaigns with significantly reduced reagent consumption compared to traditional SPR [33]. LSPR's enhanced local field sensitivity makes it particularly suitable for detecting subtle binding events involving low molecular weight compounds that produce minimal response in conventional SPR [30]. Additionally, LSPR platforms have been successfully integrated with microfluidic systems to create lab-on-a-chip devices for point-of-care therapeutic monitoring and personalized medicine applications [33]. Both technologies continue to evolve to address emerging challenges in pharmaceutical development, including the characterization of complex bispecific antibodies, antibody-drug conjugates, and gene therapy vectors.
Emerging Applications and Technological Innovations
The application landscape for SPR and LSPR continues to expand with ongoing technological innovations. Recent advances in SPR microscopy (SPRM) have enabled visualization of individual nanoparticles and single molecular binding events, opening new possibilities for digital biodetection and heterogeneous interaction analysis [34]. However, SPRM is susceptible to defocus effects that alter imaging characteristics, necessitating sophisticated focus maintenance or computational refocusing algorithms [34]. Quantitative studies have demonstrated that defocus significantly modifies the characteristic interference patterns in SPRM images, with fringe spacing increasing linearly with defocus distance – a relationship that can be exploited for rapid focus correction in long-term observations [34].
LSPR technology has witnessed revolutionary advances through the development of three-dimensional nanoplasmonic structures that dramatically enhance detection capabilities [33]. These novel substrates amplify signals by up to 1000-fold compared to conventional two-dimensional platforms, enabling detection limits in the sub-picomolar range for protein biomarkers [33]. This enhanced sensitivity facilitates new applications in early disease diagnosis through detection of low-abundance biomarkers in complex clinical samples like saliva, urine, or blood [33]. The simplified optical requirements of these enhanced LSPR platforms have enabled development of cost-effective, portable instruments potentially priced below $50,000 – dramatically increasing accessibility compared to traditional SPR systems that often exceed $200,000 [33]. Ongoing innovations continue to push the boundaries of both technologies, with integration of advanced materials, computational methods, and multiplexing capabilities further expanding their utility in binding monitoring and molecular characterization.
SPR and LSPR technologies offer complementary approaches for real-time, label-free monitoring of biomolecular interactions, each with distinct strengths and optimal application domains. SPR remains the gold standard for detailed kinetic characterization of binding interactions, providing unparalleled information about association and dissociation processes with exceptional sensitivity [31] [32]. Its well-established methodology and extensive validation across countless systems make it particularly valuable for quantitative studies requiring precise kinetic parameter determination. In contrast, LSPR technology offers advantages in spatial resolution, miniaturization potential, and reduced susceptibility to bulk effects [30]. The ongoing development of 3D nanoplasmonic structures has significantly enhanced LSPR sensitivity while reducing costs, expanding accessibility to broader research communities [33].
The selection between SPR and LSPR for specific binding monitoring applications depends on multiple factors including the required information (kinetics versus simple binding confirmation), molecular weight of analytes, available sample volume, and infrastructure constraints. SPR excels in detailed kinetic analysis and quantification of affinity parameters, while LSPR offers advantages for small molecule detection, point-of-care applications, and high-throughput screening. Both technologies continue to evolve, with SPR systems achieving higher throughput and sensitivity while LSPR platforms become more quantitative and robust. For researchers focused on quantifying interactions on sensor surfaces, both technologies provide powerful, complementary tools that can yield comprehensive understanding of binding mechanisms when applied appropriately to their optimal use cases.
The analysis of complex biological and environmental samples demands techniques that can provide comprehensive information about molecular interactions at the sensing interface. Electrochemistry-Surface Plasmon Resonance (EC-SPR) represents a powerful coupled technique that integrates the label-free, real-time monitoring capabilities of SPR with the quantitative, electrocatalytic insights of electrochemistry. This hybrid approach offers a multidimensional analytical platform particularly suited for investigating non-specific adsorption (NSA) on sensor surfaces—a critical challenge that compromises biosensor performance in complex matrices like blood, serum, and milk [1]. NSA, or fouling, refers to the accumulation of non-target species on the biosensing interface, which can mask specific signals, reduce sensitivity, and lead to false positives or negatives [1].
The fundamental strength of EC-SPR lies in its ability to provide complementary information from a single experiment. While SPR monitors mass changes and conformational rearrangements at the interface in real-time, electrochemical techniques simultaneously characterize electron transfer processes, catalytic activity, and faradaic reactions. This synergy is especially valuable for studying two-dimensional materials like graphene and transition metal dichalcogenides, which exhibit specialized physical properties that can be exploited for novel sensing principles [35]. For researchers focused on sensor surface characterization, EC-SPR provides unprecedented insights into how interfacial properties influence both fouling behavior and analytical performance.
Comparative Analysis of Techniques for NSA Investigation
The selection of an appropriate analytical technique depends heavily on the specific research questions, sample complexity, and required information dimensions. The table below compares EC-SPR with other established methods for investigating NSA and quantifying analytes on sensor surfaces.
Table 1: Technique Comparison for NSA Investigation and Sensor Analysis
| Technique | Key Advantages | Limitations for NSA Studies | Typical Applications | Quantification Capabilities |
|---|---|---|---|---|
| EC-SPR (Coupled) | Real-time monitoring of both mass changes (SPR) and electron transfer (EC); Detailed interfacial information; Evaluation of antifouling coatings [1] | Requires specialized instrumentation; Complex data interpretation; Limited commercial availability | Fundamental study of electrochemical processes at solid-liquid interfaces; Evaluation of antifouling coatings; Biosensor development [35] [1] | Dual-parameter quantification (mass, charge); Kinetic binding studies; Concentration measurements via calibration curves |
| Voltammetry with Chemometrics | High sensitivity; Cheap instrumentation; Excellent for electroactive species; Multianalyte detection with pattern recognition [36] [37] | Limited to electroactive compounds; No direct mass change information; Requires complex data processing | Simultaneous drug quantification (e.g., NSAIDs) in mixtures; Environmental monitoring; Pharmaceutical analysis [36] [37] | Nanomolar detection limits; Successful for paracetamol, diclofenac, naproxen, aspirin using ANN/PLS models [36] [37] |
| Immunochromatographic Strips | Rapid results (10 min); No sophisticated equipment needed; Suitable for on-site screening [38] | Limited multiplexing; Primarily qualitative/semi-quantitative; Limited information on NSA | Screening of specific drug classes (e.g., oxicam NSAIDs) in food safety; Point-of-care testing [38] | Visual LOD 0.25-1 ng/mL for oxicam NSAIDs in milk; Cut-off values 5-25 ng/mL [38] |
| SPR Alone | Label-free real-time binding kinetics; High-quality kinetic parameters (ka, kd, KD) [39] | No electrochemical activity information; Susceptible to bulk refractive index effects | High-throughput screening of molecular interactions; Drug discovery [39] | Affinity measurements; Concentration analysis via calibration; Hit identification in library screening [39] |
EC-SPR Experimental Workflow for NSA Evaluation
A typical EC-SPR experimental protocol for evaluating non-specific adsorption and antifouling coatings involves a systematic workflow that leverages the complementary nature of both techniques. The following diagram illustrates this integrated process:
Diagram 1: EC-SPR Experimental Workflow
Detailed Experimental Protocol
Sensor Chip Preparation and Antifouling Coating Application: The process begins with a clean SPR sensor chip integrated with a microelectrode. Researchers functionalize this surface with selected antifouling coatings, which may include polyethylene glycol (PEG) derivatives, zwitterionic polymers, cross-linked protein films, or peptide-based layers [1]. These coatings are selected based on their ability to resist fouling while maintaining the conductivity essential for electrochemical measurements and appropriate thickness for SPR sensitivity. The coating application method (e.g., self-assembled monolayers, polymer grafting, or in-situ cross-linking) must be optimized for each material to ensure uniform coverage and stability.
Baseline Establishment: Once functionalized, the sensor is mounted in the EC-SPR flow cell, and running buffer (typically PBS or HEPES) is introduced at a constant flow rate. The system establishes a stable dual-parameter baseline for both SPR angle and electrochemical current/impedance. This baseline serves as the reference point for all subsequent measurements. For electrochemical characterization, researchers often perform cyclic voltammetry in a defined potential window using redox probes like ferricyanide to verify electrode integrity and coating permeability [1].
Sample Injection and Simultaneous Monitoring: Complex samples (e.g., diluted serum, milk, or blood) are injected into the flow system while simultaneously monitoring both SPR response (reflectivity changes indicating mass adsorption) and electrochemical signals (current changes in amperometry or impedance shifts in EIS) [1]. This dual monitoring continues throughout association, dissociation, and regeneration phases. The SPR component directly tracks adsorption events in real-time, while the electrochemical component provides complementary information about how fouling impacts electron transfer rates and charge distribution at the interface.
Data Analysis: The resulting datasets require specialized analysis to deconvolute specific signals from non-specific adsorption. Researchers typically calculate fouling parameters such as the percentage of surface coverage, adsorption/desorption rates, and the impact of fouling on charge transfer resistance [1]. By comparing these parameters across different antifouling coatings, researchers can quantitatively assess coating efficacy and optimize surface chemistry for specific applications.
Research Reagent Solutions for EC-SPR Studies
Successful implementation of EC-SPR methodology requires specific materials and reagents tailored to interfacial analysis. The following table details essential research reagent solutions for EC-SPR experiments focused on NSA investigation.
Table 2: Essential Research Reagent Solutions for EC-SPR Studies
| Reagent/Category | Specific Examples | Function in EC-SPR Experiments |
|---|---|---|
| Antifouling Coatings | PEG derivatives, zwitterionic polymers, cross-linked protein films, peptide-based layers, hybrid materials [1] | Minimize non-specific adsorption; Create bioinert surfaces; Improve biosensor selectivity in complex samples |
| Surface Functionalization Reagents | Thiols (for gold surfaces), silanes (for glass/oxide), diazonium salts, NHS/EDC coupling chemistry [1] | Enable covalent attachment of antifouling coatings; Provide specific functional groups for bioreceptor immobilization |
| Redox Probes | Potassium ferricyanide, ruthenium hexamine, methylene blue [1] | Characterize electrochemical properties of modified interfaces; Monitor changes in electron transfer kinetics due to fouling |
| Complex Sample Matrices | Serum, blood, milk (often diluted), synthetic biofluids [1] | Mimic real-world conditions for NSA evaluation; Test antifouling coating efficacy under challenging conditions |
| Reference Proteins | Bovine serum albumin (BSA), fibrinogen, lysozyme [1] | Model foulant proteins for standardized NSA testing; Evaluate coating resistance to different protein types |
| Sensor Chips | Gold-coated glass slides with integrated microelectrodes, 2D material-modified chips (graphene, TMDCs) [35] | Serve as platform for EC-SPR measurements; Provide plasmonically active and electroconductive surface |
| Regeneration Solutions | Glycine-HCl (low pH), NaOH, SDS solutions [1] | Remove adsorbed species without damaging functionalized surface; Enable sensor reuse for multiple measurements |
Case Studies: EC-SPR in Sensor Surface Characterization
Investigating Antifouling Coatings for Serum Analysis
In a representative application, researchers utilized EC-SPR to evaluate novel zwitterionic polymer coatings for biosensors intended for serum analysis [1]. The SPR component monitored protein adsorption in real-time as diluted serum was injected, revealing adsorption rates of approximately 0.8 ng/cm²/min for the best-performing coating compared to >15 ng/cm²/min for unmodified gold surfaces. Simultaneously, electrochemical impedance spectroscopy measured the charge transfer resistance, demonstrating that the coating maintained efficient electron transfer (less than 20% increase in Rct) even after prolonged serum exposure. This dual confirmation proved invaluable for optimizing coating thickness to balance antifouling effectiveness with electrochemical performance.
Analyzing Drug-Protein Interactions
EC-SPR has provided unique insights into drug-protein interactions relevant to pharmaceutical development [1]. In studies monitoring nonsteroidal anti-inflammatory drug (NSAID) binding to serum proteins, SPR tracked the mass changes associated with binding events, while electrode-integrated within the flow cell monitored oxidation signals of the drug molecules. This configuration enabled researchers to distinguish between surface-bound and freely diffusing drug species, revealing interaction kinetics and binding constants that aligned with literature values but were obtained through a single, streamlined experiment. The coupled approach proved particularly valuable for detecting conformational changes in proteins upon drug binding, which manifested as shifts in both SPR angle and electrochemical current.
Data Processing in Coupled Technique Analysis
The multidimensional data generated by coupled techniques like EC-SPR and voltammetry with chemometrics requires sophisticated processing approaches. The following diagram illustrates the data processing workflow for complex voltammetric analysis of multiple NSAIDs, which shares similarities with EC-SPR data interpretation:
Diagram 2: Data Processing Workflow
For voltammetric analysis of multiple NSAIDs, researchers applied Discrete Wavelet Transform (DWT) using the db4 wavelet at the fourth decomposition level to handle the complexity and high dimensionality of the voltammograms [36] [37]. This data compression strategy extracted approximation coefficients that preserved the essential features of the original signals while significantly reducing data volume. The processed datasets were then modeled using either Partial Least Squares (PLS) regression or Artificial Neural Networks (ANN) with multilayer perceptron architecture [36]. The ANN approach demonstrated superior performance, achieving correlation values of R ≥ 0.968 for simultaneously quantifying paracetamol, diclofenac, naproxen, and aspirin in mixture solutions [36] [37]. Similar advanced data processing techniques are essential for extracting meaningful information from the complex datasets generated by EC-SPR systems.
EC-SPR stands as a powerful coupled technique that provides multidimensional insights into interfacial processes, particularly non-specific adsorption on sensor surfaces. While alternative methods like standalone voltammetry with chemometrics or immunochromatographic strips offer valuable solutions for specific applications, EC-SPR delivers unique complementary information that accelerates the development of robust biosensors for complex matrices. The continued refinement of EC-SPR instrumentation, antifouling coatings, and data processing algorithms will further establish this coupled technique as an indispensable tool for researchers investigating interfacial phenomena and developing next-generation sensors for pharmaceutical, clinical, and environmental applications.
Non-specific adsorption (NSA) is a persistent and formidable challenge in biosensing that significantly compromises sensor performance by reducing sensitivity, specificity, and reproducibility [18] [1]. NSA occurs when non-target molecules, such as proteins, indiscriminately adhere to a sensor's surface through physisorption, leading to elevated background signals that are often indistinguishable from specific binding events [18]. This phenomenon is particularly problematic in complex matrices like blood, serum, and milk, where numerous interfering substances can foul the sensing interface [1]. The consequences of NSA include false-positive readings, reduced dynamic range, and diminished detection limits, ultimately hindering the reliable application of biosensors in clinical diagnostics, drug development, and environmental monitoring [18] [40].
Material-centric approaches utilizing advanced nanomaterials and polymers have emerged as powerful strategies to combat NSA. These approaches fundamentally modify the sensor interface to create surfaces that are inherently resistant to fouling while maintaining optimal functionality for specific detection [40] [41]. This guide provides a comprehensive comparison of these coating strategies, detailing their performance characteristics, experimental validation protocols, and practical implementation for researchers developing next-generation biosensing platforms.
Coating Mechanisms and Material Classifications
Coatings to prevent NSA operate primarily through two fundamental mechanisms: creating a physical barrier that prevents foulants from reaching the sensor surface, and engineering surface chemistries that exhibit minimal interaction with non-target molecules [18] [1]. These mechanisms can be achieved through various material strategies, broadly categorized into passive and active methods.
Passive methods rely on creating a thin, hydrophilic, and non-charged boundary layer that thwarts protein adsorption through thermodynamic principles [18]. These include traditional blocking proteins like bovine serum albumin (BSA) and casein, as well as more advanced chemical coatings such as self-assembled monolayers (SAMs) and polymer films [18] [40]. Active methods dynamically remove adsorbed molecules post-functionalization by generating surface forces that shear away weakly adhered biomolecules, typically using electromechanical or acoustic transducers, or hydrodynamic fluid flow [18].
Table 1: Classification of NSA-Reducing Coating Strategies
| Coating Category | Mechanism of Action | Key Materials | Primary Applications |
|---|---|---|---|
| Polymer Films | Creates a hydrated physical barrier that sterically hinders fouling | PEG, PSS, Polyurethanes, Acrylics | Optical biosensors, Electrochemical sensors, Microfluidics |
| Nanomaterial Coatings | Enhances evanescent field coupling; provides high surface area for functionalization | Gold nanoparticles, Quantum Dots, Graphene, MOFs | SPR/LSPR sensors, Optical fiber sensors, EC-SPR platforms |
| Self-Assembled Monolayers (SAMs) | Forms ordered molecular structures that minimize interfacial interactions | Alkanethiols on gold, Silanes on glass | Patterned biosensors, Immunosensors |
| Blocking Proteins | Physically occupies vacant surface sites | BSA, Casein, Milk proteins | ELISA, Western blotting, Routine immunoassays |
The following diagram illustrates the core mechanisms through which nanomaterials and polymers mitigate non-specific adsorption on sensor surfaces:
Diagram: Material-centric anti-NSA mechanisms create barriers through hydration, steric hindrance, charge repulsion, and morphology control.
Comparative Performance Analysis of Coating Materials
Polymer-Based Coating Systems
Polymer coatings represent one of the most extensively studied approaches for mitigating NSA, with their performance heavily dependent on chemical composition, thickness, and application method. The effectiveness of various polymer systems has been quantitatively evaluated through multiple experimental studies.
Table 2: Performance Comparison of Polymer-Based Anti-NSA Coatings
| Polymer Type | Coating Method | Experimental NSA Reduction | Limit of Detection Improvement | Key Advantages |
|---|---|---|---|---|
| PSS (Poly(styrene sulfonic acid) sodium salt) | Self-assembly | ~300-fold reduction in QD adsorption [40] | CRP detection: 1.3 ng/mL [40] | Dense negative charge; simple processing |
| TSPP (sulfonated porphyrin) | Self-assembly | ~400-fold reduction in QD adsorption [40] | CRP detection: 5.2 ng/mL [40] | Multiple sulfonate groups; strong charge repulsion |
| TSPP/PSS Hybrid | Sequential self-assembly | Superior to individual components [40] | CRP detection: 0.69 ng/mL [40] | Combines high charge density with minimized FRET |
| Polyurethane | Spray/dip coating | Industry standard for durability [42] [43] | Not specifically quantified for biosensing | Exceptional mechanical properties; chemical resistance |
| Acrylics | Spin coating, Dip coating | High weatherability and color retention [44] | Varies by formulation | Low VOC; excellent adhesion; cost-effective |
Recent research on sequential self-assembly of TSPP and PSS demonstrates that hybrid approaches can yield superior performance. This strategy achieved a remarkable limit of detection (LOD) of 0.69 ng/mL for C-reactive protein (CRP), representing a 1.9-fold and 7.5-fold improvement over PSS-only and TSPP-only modified surfaces, respectively [40]. The enhanced performance is attributed to the combination of high negative charge density with optimized spacing that minimizes fluorescence resonance energy transfer (FRET) between TSPP and quantum dot labels.
Nanomaterial-Enhanced Coating Systems
Nanomaterials impart unique advantages for NSA reduction through their high specific surface area, quantum confinement effects, and capacity for creating sophisticated surface morphologies that minimize non-specific interactions.
Table 3: Performance Comparison of Nanomaterial-Based Anti-NSA Coatings
| Nanomaterial | Coating Method | Sensor Platform | NSA Reduction Efficacy | Unique Properties |
|---|---|---|---|---|
| Gold Nanoparticles | Self-assembly, Sputtering | SPR, LSPR | High (size-dependent) [45] | Tunable plasmonic properties; easy functionalization |
| Quantum Dots | Spin coating, Self-assembly | Fluorescence immunosensors | Enabled ultra-sensitive detection [40] | High photoluminescence; photobleaching resistance |
| TiO₂ Nanoparticles | Atomic Layer Deposition | Optical fiber sensors | Phase-dependent performance [45] | High refractive index; photocatalytic properties |
| SiO₂ Nanoparticles | Dip coating, Spray coating | Multiple platforms | Improved by surface functionalization [41] | Excellent hydrophilicity; tunable surface chemistry |
| MOFs (Metal-Organic Frameworks) | Electrochemical deposition | EC-SPR platforms | High for small molecules [41] | Ultra-high porosity; molecular sieving capabilities |
The performance of nanomaterial coatings is highly dependent on precise control of physical parameters. For example, noble metal nanoparticles exhibit size-dependent optical properties, with particles below 20 nm demonstrating significantly different magnetic, dielectric, and optical characteristics compared to their bulk counterparts [45]. Similarly, TiO₂ nanoparticles undergo phase-dependent behavior, with anatase becoming more stable than rutile when particle size decreases below approximately 14 nm, directly impacting their refractive index and band gap properties relevant to sensing applications [45].
Experimental Protocols for Coating Evaluation
Standardized Methodologies for NSA Quantification
Robust evaluation of anti-NSA coatings requires standardized protocols and quantitative metrics to enable cross-comparison between different material systems. The following experimental workflows represent best practices for coating validation:
Protocol 1: Fluorescence-Based NSA Quantification
- Surface Preparation: Clean substrate (e.g., glass slide) with piranha solution, followed by extensive rinsing with deionized water [40].
- Coating Application: Functionalize surface with target material (e.g., PSS, TSPP) via self-assembly, dip-coating, or spin-coating using optimized parameters [40] [41].
- Exposure to Fluorophores: Incubate coated surface with fluorescent probes (e.g., aqueous quantum dots) for standardized duration under controlled conditions [40].
- Signal Measurement: Quantify fluorescence intensity using calibrated instrumentation; compare against non-functionalized control surfaces [40].
- Data Analysis: Calculate NSA reduction factor as the ratio of fluorescence intensity on control vs. functionalized surfaces [40].
Protocol 2: SPR-Based Binding Kinetics Analysis
- Sensor Functionalization: Deposit nanocoating on SPR chip using appropriate method (e.g., SAM formation, polymer grafting) [1].
- Baseline Establishment: Flow buffer solution to establish stable baseline signal [1].
- Foulant Challenge: Introduce complex media (e.g., serum, plasma) or protein solutions to assess non-specific binding [1].
- Regeneration: Apply regeneration buffer to remove weakly bound species [1].
- Data Interpretation: Quantify NSA by measuring response units (RU) retained after regeneration phase; compare with specific binding signals [1].
The experimental workflow for evaluating coating efficacy typically follows a systematic path from substrate preparation to data interpretation, as illustrated below:
Diagram: Experimental workflow for evaluating anti-NSA coatings progresses from substrate preparation to data analysis.
Advanced Characterization Techniques
Comprehensive evaluation of anti-NSA coatings necessitates multi-modal characterization to correlate physical properties with functional performance:
- Surface Topography: Atomic Force Microscopy (AFM) and Scanning Electron Microscopy (SEM) provide nanoscale resolution of coating morphology and uniformity [46] [41].
- Chemical Composition: X-ray Photoelectron Spectroscopy (XPS) and Fourier-Transform Infrared Spectroscopy (FTIR) verify surface chemistry and successful functionalization [46].
- Hydrophilicity Assessment: Contact angle measurements quantify surface energy and wettability, key parameters influencing protein adsorption [1].
- Thickness Profilometry: Ellipsometry and profilometry precisely measure coating thickness, critical for optimizing optical and electrochemical sensors [18] [41].
- Electrochemical Characterization: For conductive coatings, electrochemical impedance spectroscopy (EIS) and cyclic voltammetry assess electron transfer rates and interface properties [1].
The Scientist's Toolkit: Essential Materials and Reagents
Successful implementation of anti-NSA coatings requires access to specialized materials, instruments, and analytical capabilities. The following toolkit summarizes key resources for researchers developing and evaluating these advanced material systems:
Table 4: Essential Research Toolkit for Anti-NSA Coating Development
| Category | Specific Items | Primary Function | Representative Examples |
|---|---|---|---|
| Substrate Materials | Glass slides, Gold chips, Silicon wafers, Optical fibers | Foundation for coating development | Sail Brand glass slides [40] |
| Polymer Coating Reagents | PSS, PEG derivatives, Polyurethane precursors, Acrylic emulsions | Create anti-fouling surface layers | Poly(styrene sulfonic acid) sodium salt [40] |
| Nanomaterials | Gold nanoparticles, Quantum dots, SiO₂ nanoparticles, Graphene oxide | Enhance sensing properties and reduce NSA | CdSe/ZnS quantum dots [40] |
| Surface Characterization Instruments | AFM, SEM, Ellipsometer, Contact angle goniometer | Quantify physical properties of coatings | Various commercial systems [46] [41] |
| Bioanalytical Assessment Tools | SPR systems, Fluorescence scanners, Electrochemical workstations | Evaluate NSA and sensing performance | EC-SPR platforms [1] |
| Blocking Agents | BSA, Casein, Milk proteins, Synthetic blocking peptides | Traditional NSA reduction for comparison | Bovine serum albumin [18] |
Material-centric approaches utilizing advanced polymers and nanomaterials represent a powerful strategy for addressing the persistent challenge of non-specific adsorption in biosensors. The comparative analysis presented in this guide demonstrates that both material classes offer distinct advantages, with polymer systems typically providing robust, cost-effective protection, while nanomaterial coatings enable enhanced sensitivity and unique sensing mechanisms through their distinctive optical, electrical, and structural properties.
Future developments in this field will likely focus on multi-functional coating systems that combine the strengths of both material classes, such as polymer-nanoparticle composites that offer synergistic anti-fouling and sensing capabilities [45]. Additionally, the growing emphasis on sustainability is driving research toward bio-based polymers and green synthesis pathways for nanomaterials [43] [47] [44]. Emerging trends include the application of machine learning and molecular simulations to design next-generation coating materials with optimized anti-NSA properties, potentially revolutionizing the development timeline for high-performance biosensing interfaces [1].
As biosensor technologies continue to evolve toward point-of-care applications and complex sample analysis, the critical importance of effective NSA control cannot be overstated. The material-centric approaches detailed in this guide provide a robust foundation for developing biosensors with the reliability, sensitivity, and specificity required for advanced diagnostic and research applications.
Strategies for NSA Mitigation: Antifouling Coatings and Surface Engineering
The nonspecific adsorption (NSA) of biomolecules onto sensor surfaces is a fundamental challenge in diagnostic and drug development, leading to signal noise, reduced sensitivity, and unreliable data [48]. Passive antifouling methods, which prevent this initial, unwanted adhesion, are therefore critical for the performance of biosensors and other analytical platforms. Unlike active methods that release biocides or use enzymatic action to eliminate fouling, passive strategies create a thermodynamically unfavorable or physically impermeable barrier that resists the adsorption of proteins, cells, and other contaminants [49] [50]. This guide objectively compares the performance of advanced passive antifouling materials, including synthetic polymers, bioinspired peptides, and supramolecular blocking agents, providing researchers with quantitative data and detailed experimental protocols to inform their selection for sensor surface functionalization.
Performance Comparison of Passive Antifouling Materials
The efficacy of passive antifouling materials is typically quantified by their ability to reduce the adhesion of model proteins and bacterial cells compared to control surfaces. The following tables summarize key performance metrics for the major classes of materials, providing a basis for direct comparison.
Table 1: Antifouling Performance of Hydrophilic Polymer Brushes
| Polymer Material | Grafting Method | Substrate | Test Foulant | Reduction vs. Control | Key Mechanism |
|---|---|---|---|---|---|
| Polyethylene Glycol (PEG) [48] | Drop coating/Spin coating | Glass, SPR chips | E. coli, S. aureus, P. aeruginosa | Up to 99% suppression over 7 days | Osmotic repulsion from tightly bound water layer |
| Polyethylene Glycol (PEG) [48] | Polymer brush (ATRP) | Glass | E. coli | ~80% reduction after 30 min | Osmotic repulsion |
| Poly(oxazoline) (POZ) [48] | Surface-initiated ATRP | Various | Bacteria | Comparable to PEG | Hydrogen bond acceptor, neutral charge |
| Zwitterionic Polymers [48] | Surface-initiated ATRP | Various | Bacteria | Up to 99% reduction (superior to PEG) | Strong electrostatic hydration |
Table 2: Performance of Low-Surface-Energy and Supramolecular Materials
| Material Class | Specific Composition | Test Conditions | Key Performance Metric | Proposed Mechanism |
|---|---|---|---|---|
| Synergistic Resistance-Release [51] | Cyclodextrin/PDMS Supramolecular Complex | BSA solution, 550 L·m⁻²·h⁻¹ flux, 60 rpm stirring | 14.2% flux decline | Dynamic hydrophilic/LSE microdomains prevent foulant accumulation |
| Low-Surface-Energy Coating [50] | P-RuSe₂ NPs in hydrophobic layer (VR coating) | 180-day marine field test | 86.22% reduced biofouling vs. control | Passive reduction of initial organism adhesion |
| Fluoropolymer [52] | PFA, PTFE | Pancake batter release test | Pull-off force: <3 kPa | Inherent non-stick, low surface energy |
Experimental Protocols for Key Systems
To ensure reproducibility and facilitate the adoption of these materials, detailed methodologies for the preparation and testing of two high-performing systems are outlined below.
Protocol: Supramolecular Cyclodextrin/PDMS Antifouling Layer
This protocol describes the creation of a dynamic antifouling surface with synergistic resistance-release properties, suitable for applications under low tangential flow [51].
1. Synthesis of CD/PDMS Polyrotaxanes (PRs):
- Materials: Polydimethylsiloxane (PDMS, 5000 Da), γ-Cyclodextrin (γ-CD), N,N-Dimethylformamide (DMF).
- Procedure: Dissolve γ-CD and PDMS in DMF solvent. Stir the mixture at 60°C for 48 hours to facilitate the threading of γ-CD molecules onto the linear PDMS chain. Control the molar ratio of CD to PDMS units (rCD:PDMS) to optimize the dynamic mobility of the complex. Precipitate the resulting CD/PDMS poly(pseudo)rotaxane (PPR) product in cold diethyl ether and purify via filtration.
- Grafting: The PPRs are grafted onto the target membrane surface via the PDMS end groups, converting them into stable polyrotaxanes (PRs) where the CDs cannot detach.
2. Antifouling Performance Evaluation:
- Foulant Solution: Prepare a 1 g/L solution of Bovine Serum Albumin (BSA) in a suitable buffer (e.g., phosphate-buffered saline).
- Filtration Test: Conduct dead-end or cross-flow filtration using the modified membrane.
- Conditions: Set the initial water flux to 550 L·m⁻²·h⁻¹. Apply constant stirring at 60 rpm to simulate low tangential flow.
- Data Analysis: Measure the flux decline over time. The flux decline ratio is calculated as:
(1 - J/J₀) × 100%, where J₀ is the initial flux and J is the flux at time t. A lower flux decline indicates superior antifouling performance.
The following workflow diagram illustrates the preparation and testing process for the CD/PDMS supramolecular antifouling layer:
Protocol: Zwitterionic Polymer Brushes via ATRP
This protocol details the creation of a highly effective nonfouling surface using zwitterionic polymer brushes, known for their exceptional hydration capacity [48].
1. Surface Initiation:
- Materials: Dopamine-based ATRP initiator, Tris buffer, ethanol.
- Substrate Preparation: Clean the substrate (e.g., gold, silicon, glass) thoroughly with ethanol and plasma treatment.
- Procedure: Immerse the substrate in a solution of the bromine-terminated ATRP initiator. For silicon or glass, a silane-based initiator is used. The initiator forms a self-assembled monolayer, providing a covalently anchored base for polymer growth.
2. Surface-Initiated Atom Transfer Radical Polymerization (SI-ATRP):
- Materials: Zwitterionic monomer, Cu(I)Br catalyst, ligand.
- Procedure: Prepare a degassed mixture of the zwitterionic monomer, catalyst, and ligand in a water/solvent solution. Place the initiator-modified substrate into the reaction mixture. Allow polymerization to proceed for a controlled period (e.g., 1-24 hours) under an inert atmosphere to control brush length and density.
- Termination: Remove the substrate and rinse thoroughly with deionized water to terminate the reaction and remove any physisorbed catalyst or monomer.
3. Bacterial Adhesion Assay:
- Bacterial Strains: Use relevant models like Escherichia coli (Gram-negative) and Staphylococcus aureus (Gram-positive).
- Procedure: Incubate the coated substrates in a bacterial suspension for a set period (e.g., 30 minutes to 24 hours).
- Analysis: Gently rinse the substrates to remove non-adherent cells. Stain the adherent bacteria and count them using fluorescence microscopy or quantify via colony-forming unit (CFU) counts. The percentage reduction is calculated versus an uncoated control.
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Reagents for Passive Antifouling Research
| Reagent / Material | Function / Role | Example Application |
|---|---|---|
| Cyclodextrin (CD) & Polydimethylsiloxane (PDMS) [51] | Forms dynamic, threaded supramolecular complexes that create heterogeneous antifouling microdomains. | Building synergistic resistance-release surfaces for filtration membranes. |
| Zwitterionic Monomers [48] | Polymerize to form brushes that bind water molecules strongly via electrostatic hydration, creating a physical and energetic barrier to adsorption. | Creating ultra-low fouling surfaces for biosensors and medical implants. |
| PEG-based ATRP Initiator [48] | Provides a surface-tethered starting point for growing polymer brushes with controlled density and thickness via Atom Transfer Radical Polymerization. | Grafting PEG and other polymer brushes onto metal oxide surfaces. |
| Dopamine-based ATRP Initiator [48] | A versatile initiator that adheres to a wide range of organic and inorganic substrates, enabling surface-initiated polymerization. | Modifying diverse sensor surfaces (polymers, metals, oxides) with antifouling brushes. |
| Bovine Serum Albumin (BSA) [51] | A model protein foulant used for initial, standardized evaluation of a surface's resistance to nonspecific protein adsorption. | Quantitative filtration flux decline tests. |
For researchers characterizing NSA on sensor surfaces, the choice of a passive antifouling strategy is a critical determinant of data reliability. The quantitative data presented here demonstrates that while traditional materials like PEG remain effective, emerging zwitterionic polymers and dynamic supramolecular materials can offer superior performance. The decision matrix should integrate material performance with practical constraints: zwitterionic brushes grafted via ATRP may provide the gold standard for maximum fouling resistance in static or low-flow sensor systems, whereas CD/PDMS polyrotaxanes offer a compelling solution for microfluidic devices where dynamic flow can be harnessed for enhanced foulant release. Integrating these advanced passive layers is paramount for developing next-generation sensors with unparalleled accuracy in complex biological environments.
The precise removal of nonspecific adsorption (NSA) from sensor surfaces is a critical challenge in biosensor development and drug discovery. Residual analyte or contaminant molecules can significantly compromise signal integrity, leading to inaccurate data and false conclusions. Among the various techniques developed to address this issue, electromechanical and hydrodynamic shearing have emerged as two prominent physical methods. These techniques utilize distinct fundamental principles—electrically induced forces and fluid flow-induced stresses, respectively—to achieve surface cleaning without damaging delicate sensor substrates. This guide provides an objective comparison of their performance, supported by experimental data and detailed protocols, to inform researchers and development professionals in selecting the appropriate method for their specific application needs.
Fundamental Principles and Mechanisms
Hydrodynamic Shearing
Hydrodynamic shearing relies on the mechanical force exerted by a moving fluid to dislodge and remove particles or molecules adsorbed onto a surface. When a fluid flows over a surface, it creates a shear stress (τ) at the solid-liquid interface. This stress is a function of the fluid's dynamic viscosity (μ) and the velocity gradient (du/dy) perpendicular to the surface, as described by the equation for a Newtonian fluid: τ = μ (du/dy) [53] [54].
The effectiveness of removal is directly correlated to the magnitude of this shear stress. Computational Fluid Dynamics (CFD) simulations are often employed to model the shear stress distribution across a surface, revealing that stress is highest near the boundaries of the flow path, such as the walls of an extrusion nozzle or a flow cell [54]. Experimental studies have demonstrated a clear negative correlation between applied shear stress and surface porosity; as shear stress increases, the presence of adsorbed layers or structures diminishes, effectively cleaning the surface [54]. For instance, in applications involving graphene-oxide aerogels, a shear stress threshold of approximately 338 Pa was identified, above which surfaces became entirely non-porous, indicating effective clearance of porous obstructions [54].
Electromechanical Removal
Electromechanical methods harness electrical phenomena to generate mechanical forces that repel or remove adsorbed species. A key mechanism involves the use of electrospun nanofiber membranes, which can be engineered to possess specific charge characteristics [55]. During the electrospinning process, functional polymers gain a net charge, and the resulting nanofibers can exhibit either surface or volume charge distributions [55].
These charges create an electrostatic environment that can inhibit the adsorption of molecules or facilitate their release. Furthermore, such charged membranes can be integrated into triboelectric nanogenerators (TENGs), where mechanical contact and separation generate a transient electrical output. The voltage polarity and molecular orientation on the nanofiber surface, controlled by the electrospinning voltage, can be tuned to enhance this output, which in turn can exert electromechanical forces on nearby adsorbed species [55]. For example, a negatively electrospun polyvinyl chloride (PVC) nanofiber membrane was shown to achieve a triboelectric output of 12 V, higher than its positively spun counterpart, demonstrating the ability to control surface properties for active functionality [55].
Table 1: Fundamental Characteristics of Removal Methods
| Feature | Hydrodynamic Shearing | Electromechanical Removal |
|---|---|---|
| Primary Force | Fluid shear stress (τ) | Electrostatic and triboelectric forces |
| Governing Equation | τ = μ (du/dy) | Dependent on material charge density and TENG output |
| Key Controlling Parameter | Flow rate, fluid viscosity, channel geometry | Applied voltage, polymer functional groups, voltage polarity |
| Typical Measured Output | Shear stress (Pa) | Surface potential (V) or charge density |
Experimental Performance Comparison
Efficiency and Key Performance Metrics
Direct comparative studies on sensor surfaces are limited in the provided search results; however, data from related applications such as surface structuring and filtration provide valuable performance insights.
Hydrodynamic shearing demonstrates quantifiable performance thresholds. In controlled studies of graphene-based aerogel fabrication, the surface porosity—a proxy for material adherence—was directly controlled by shear stress. Applying a shear stress of ~316 Pa resulted in a porous surface, while stresses above ~338 Pa produced a non-porous, cleared surface. In a different setup using calcium ion-crosslinked inks, the transition threshold was higher, between ~500 Pa and ~557 Pa, indicating that the required shear stress is highly dependent on the adhesive strength of the surface material [54].
Electromechanical methods show efficacy in altering surface interactions through charge. The performance of electrospun nanofiber membranes in air filtration, which relies on capturing particles via electrostatic interactions, is largely dependent on their charge properties. The electron-withdrawing capacity of functional groups in the polymer determines the initial charge density, which directly enhances filtration efficiency [55]. The ability to tune the voltage polarity during electrospinning to regulate molecular orientation is a key strategy for boosting the output of TENGs, with a demonstrated maximum output voltage of 12 V for a negatively electrospun PVC membrane [55]. This electrical output can be harnessed to create repulsive forces against adsorbed species.
Table 2: Summary of Experimental Performance Data
| Method | Experimental Context | Key Performance Metric | Result | Reference |
|---|---|---|---|---|
| Hydrodynamic Shearing | Porosity control of GO inks with 40 mM AA | Transition Shear Stress (Porous to Non-porous) | ~316 Pa to ~338 Pa | [54] |
| Hydrodynamic Shearing | Porosity control of GO inks with 6 mM Ca²⁺ | Transition Shear Stress (Porous to Non-porous) | ~500 Pa to ~557 Pa | [54] |
| Electromechanical | Electrospun PVC Nanofiber Membrane | Triboelectric Nanogenerator (TENG) Output | 12 V (max) | [55] |
Advantages and Limitations
Hydrodynamic Shearing:
- Advantages: The mechanism is well-understood and easily modeled with CFD. It allows for uniform application over large or complex surface areas within a flow cell and is a non-contact method for the surface being cleaned, minimizing risk of physical damage [53] [54].
- Limitations: Its effectiveness can be limited in microscopic pores or complex nanostructures where fluid flow is restricted. The requirement for fluid handling systems (pumps, reservoirs) can increase the complexity of the setup. High shear stresses, while effective for cleaning, could potentially damage very fragile sensor coatings [54].
Electromechanical Removal:
- Advantages: This method can generate significant forces at the micro/nano scale without requiring bulk fluid movement. The surface properties can be tuned for specific applications by selecting different polymers and electrospinning parameters. It shows promise for integrated, self-powered systems using TENGs [55].
- Limitations: The efficiency is highly dependent on the environmental conditions, such as humidity, which can dissipate surface charges. The process may require the fabrication and integration of specialized membranes. The long-term stability of the charge on functionalized surfaces can be a concern for repeated use [55].
Detailed Experimental Protocols
Protocol for Hydrodynamic Shearing Analysis
This protocol outlines a method to quantify the effect of hydrodynamic shear on a surface, adapted from studies on surface porosity [54].
1. Materials and Equipment:
- Flow Cell System: A precision flow cell with well-defined geometry (e.g., rectangular channel).
- Fluid Delivery System: A high-precision syringe or peristaltic pump capable of generating a range of stable flow rates.
- Test Fluid: A solution with known and constant viscosity (e.g., deionized water or a buffer solution).
- Surface Characterization Instrument: Scanning Electron Microscope (SEM) or an optical surface profiler.
- Computational Fluid Dynamics (CFD) Software: For simulating shear stress distribution (e.g., ANSYS Fluent, COMSOL Multiphysics).
2. Procedure: a. Surface Preparation: Prepare the sensor surface with a standardized layer of the analyte or contaminant to be removed. b. Experimental Setup: Mount the surface securely in the flow cell. Connect the fluid delivery system and ensure there are no leaks. c. CFD Simulation: Model the flow cell geometry and the intended flow rates in the CFD software to calculate the wall shear stress distribution across the sensor surface. This step predicts the shear stress values before the experiment. d. Shearing Process: For each experimental run, perfuse the test fluid through the flow cell at a predetermined, constant flow rate for a set duration (e.g., 10 minutes). e. Post-treatment Analysis: Carefully disassemble the flow cell and dry the sensor surface without causing contamination. Image the surface using SEM or profile it with the optical profiler. f. Quantification: Analyze the images to quantify the remaining surface coverage or porosity. Calculate the corresponding average shear stress from the CFD results for each flow rate. g. Data Correlation: Plot the residual surface coverage or porosity against the applied shear stress to establish a clearance curve for the specific surface-analyte system.
Protocol for Electromechanical Removal via Electrospun Membranes
This protocol describes the creation and testing of a functionalized surface for electromechanical removal, based on research into electrospun nanofiber membranes [55] [56].
1. Materials and Equipment:
- Electrospinning Apparatus: High-voltage power supply, syringe pump, metallic needle (spinneret), and grounded collector.
- Polymer Solution: A solution of a suitable polymer (e.g., Polyvinyl Chloride - PVC) in an appropriate solvent (e.g., Tetrahydrofuran - THF, Dimethylformamide - DMF) [56].
- Charge Characterization Tool: Atomic Force Microscope (AFM) with a Kelvin Probe Force Microscopy (KPFM) mode to measure surface potential.
- Triboelectric Testing Setup: Equipment to measure the voltage/current output of the membrane under cyclic contact-separation.
2. Procedure: a. Membrane Fabrication: - Load the polymer solution into a syringe fitted with a metallic needle. - Set the syringe pump to a constant flow rate (e.g., 1.0 mL/h). - Apply a high voltage (e.g., 15-20 kV) between the needle and the collector, which is placed at a fixed distance (e.g., 15 cm). - Collect the nanofibers formed on the collector substrate. The polarity of the applied voltage (positive or negative) will influence the molecular orientation and charge properties of the resulting membrane [55]. b. Charge Characterization: Use AFM-KPFM to map the surface potential and identify charge distribution patterns (e.g., surface charges, charges at fiber cross-points) on the newly produced nanofiber membrane [55]. c. Triboelectric Output Measurement: Integrate the membrane into a TENG setup. Measure the open-circuit voltage and/or short-circuit current generated when the membrane is subjected to periodic mechanical contact. d. Removal Efficiency Test: Expose the charged membrane to a solution containing the target analyte. After a set adsorption period, activate the TENG mechanism or utilize the inherent surface charge. Quantify removal efficiency by comparing the sensor signal or surface analyte concentration before and after the electromechanical treatment.
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 3: Key Reagents and Materials for Shearing Experiments
| Item Name | Function/Application | Key Characteristics |
|---|---|---|
| Precision Syringe Pump | To generate precise and steady flow for hydrodynamic shearing. | High flow rate accuracy, minimal pulsation. |
| CFD Software (e.g., ANSYS) | To model flow fields and predict shear stress distribution on surfaces. | Capable of laminar and turbulent flow simulation. |
| Electrospinning Apparatus | To fabricate functional nanofiber membranes for electromechanical studies. | High-voltage DC power supply, programmable syringe pump. |
| Polyvinyl Chloride (PVC) | A versatile polymer for creating electrospun membranes. | High chemical resistance, ease of electrospinning, cost-effective [56]. |
| Atomic Force Microscope (AFM) | To characterize surface topography and map surface charge/potential. | Kelvin Probe Force Microscopy (KPFM) capability. |
| Tetrahydrofuran (THF) | A common solvent for dissolving PVC for electrospinning [56]. | High volatility, good solubility for PVC. |
Conceptual Workflows and Signaling Pathways
The following diagram illustrates the logical sequence and key decision points in selecting and applying these removal methods within a research workflow.
Research Workflow for Removal Method Selection
The diagram above outlines a decision-making framework. The choice between Hydrodynamic Shearing (blue) and Electromechanical (red) methods depends on the initial assessment. Hydrodynamic shearing is suitable for stable fluid environments and well-modeled flow cells, whereas the electromechanical approach is preferred for systems with charge-sensitive components or where integrated, functionalized membranes are desired. Both paths converge on the critical evaluation of clearance efficacy.
Designing Effective Evaluation Protocols for Complex Real-World Samples
Non-specific adsorption (NSA) represents a critical barrier to the widespread adoption and reliability of biosensors, particularly when deployed for analysis in complex matrices such as blood, serum, and milk [1]. NSA refers to the accumulation of non-target sample components (foulants) on the biosensing interface, which can severely compromise analytical characteristics including signal stability, selectivity, sensitivity, and accuracy [1]. The development of robust evaluation protocols is therefore essential for characterizing NSA and validating the efficacy of antifouling strategies, ultimately determining the real-world viability of biosensing platforms.
This guide provides a comparative evaluation of experimental approaches for quantifying NSA, with a specific focus on methodologies applicable to sensors utilizing combined electrochemical-surface plasmon resonance (EC-SPR) detection. These coupled systems are particularly valuable for NSA evaluation as they enable larger detection ranges, improved spatial resolution, and more detailed information on interfacial events compared to single-method approaches [1].
Comparative Analysis of NSA Evaluation Methods
The effectiveness of an NSA evaluation protocol is intrinsically linked to the sensitivity of the method employed. A multi-technique approach often provides a more comprehensive dimension of NSA than any single method [1]. The following section compares prominent techniques used for quantifying NSA on sensor surfaces.
Table 1: Comparison of Primary Techniques for NSA Evaluation on Sensor Surfaces
| Technique | Fundamental Principle | Key Measurable Outputs for NSA | Typical Experimental Readouts | Advantages | Limitations |
|---|---|---|---|---|---|
| Surface Plasmon Resonance (SPR) | Measures changes in refractive index at a metal surface [1]. | Change in resonance angle or response units (RU) due to mass adsorption [1]. | Sensorgrams (Response vs. Time); Angular or Wavelength Spectra. | Label-free, real-time kinetic data; High surface sensitivity. | Difficult to distinguish specific binding from NSA without controls; Limited penetration depth of evanescent field. |
| Electrochemical (EC) Methods | Monitors changes in electron transfer kinetics at the electrode-solution interface [1]. | Signal drift; Changes in charge transfer resistance (Rct), capacitance, or faradaic current [1]. | Cyclic Voltammograms; Electrochemical Impedance Spectra; Amperometric/Potentiometric I-t curves. | Highly sensitive to interfacial changes; Can probe passivation effects on electron transfer. | Signal can be influenced by factors unrelated to fouling (e.g., pH, ionic strength). |
| Coupled EC-SPR | Simultaneously monitors electrochemical parameters and SPR response [1]. | Correlated signal drift in both EC and SPR channels; Dissimilar responses to provide interfacial details [1]. | Synchronized sensorgrams and electrochemical data (e.g., EIS, CV). | Provides complementary information on interfacial events; Broader detection range; Can deconvolute complex binding/adsorption events. | Experimentally more complex; Requires specialized instrumentation and customized surfaces. |
Detailed Experimental Protocols
This section outlines detailed, actionable methodologies for key experiments cited in the comparative analysis.
Protocol for NSA Evaluation using SPR
This protocol is designed to quantify the extent of fouling from complex samples onto a sensor surface.
- Sensor Surface Preparation: A bare gold sensor chip (or one with a pre-immobilized bioreceptor for specific binding studies) is mounted in the SPR instrument. The flow system is primed with running buffer (e.g., PBS, pH 7.4).
- Baseline Establishment: A stable baseline is established by flowing the running buffer over the sensor surface at a constant flow rate (e.g., 20-30 µL/min) until a stable signal is achieved.
- Sample Injection & NSA Monitoring: The complex sample (e.g., 1-10% serum in buffer, undiluted milk supernatant) is injected over the sensor surface for a defined period (e.g., 10-30 minutes). The increase in SPR response (in Response Units, RU) is recorded in real-time, representing the total adsorption (specific + non-specific).
- Rinsing & Signal Stabilization: The running buffer is reintroduced to rinse away loosely adsorbed materials. The remaining change in SPR response after stabilization is recorded as the total adsorbed mass.
- Control for Specific Binding (If applicable): For surfaces with bioreceptors, a specific inhibitor or a non-target analyte can be used to differentiate between specific binding and NSA. Alternatively, the response on a non-functionalized reference channel is subtracted.
- Data Analysis: The final, stabilized response (in RU) after rinsing is used to quantify NSA. A response of 1000 RU is approximately equivalent to a surface coverage of 1 ng/mm² [1].
Protocol for NSA Evaluation using Electrochemical Impedance Spectroscopy (EIS)
This protocol assesses the fouling-induced passivation of an electrode surface by monitoring electron transfer resistance.
- Electrode Preparation: A working electrode (e.g., gold, glassy carbon) is cleaned and characterized in a redox probe solution (e.g., 5 mM K₃[Fe(CN)₆]/K₄[Fe(CN)₆] in PBS).
- Initial EIS Measurement: An EIS spectrum is recorded at the open-circuit potential or a defined DC potential. A small AC amplitude (e.g., 10 mV) is applied across a frequency range (e.g., 0.1 Hz to 100 kHz). The initial charge transfer resistance (Rct,initial) is extracted by fitting the data to a Randles equivalent circuit.
- Fouling Phase: The electrode is incubated in the complex sample (e.g., serum, milk) for a predetermined time (e.g., 30 minutes) under static or gentle agitation conditions.
- Rinsing: The electrode is gently rinsed with running buffer to remove non-adsorbed components.
- Final EIS Measurement: The EIS measurement is repeated in the same redox probe solution, and the new charge transfer resistance (Rct,final) is determined.
- Data Analysis: The percentage increase in charge transfer resistance is calculated as a metric for fouling: % Increase in Rct = [(Rct,final - Rct,initial) / Rct,initial] × 100. A higher value indicates greater surface passivation due to NSA.
Workflow for NSA Evaluation
The following diagram illustrates the general experimental workflow for evaluating NSA and antifouling coatings, integrating both SPR and EC methodologies.
The Scientist's Toolkit: Essential Reagents & Materials
Successful execution of NSA evaluation protocols requires specific reagents and materials. The following table details key components for a typical biosensor antifouling research laboratory.
Table 2: Essential Research Reagent Solutions for NSA Studies
| Item | Function/Description | Application Example |
|---|---|---|
| Gold Sensor Chips | Substrate for SPR measurements; provides a surface for functionalization with antifouling coatings and bioreceptors [1]. | Used as the foundational sensing element in SPR and EC-SPR biosensors. |
| Redox Probes | Electroactive molecules used to probe electron transfer efficiency at an electrode surface. | K₃[Fe(CN)₆]/K₄[Fe(CN)₆] is used in EIS to monitor fouling-induced changes in charge transfer resistance (Rct) [1]. |
| Antifouling Polymers | Materials designed to minimize NSA by creating a hydrophilic, neutral barrier. | Polyethylene glycol (PEG), zwitterionic polymers, and hydrogels are coated on sensor surfaces to resist protein adsorption [1]. |
| Complex Sample Matrices | Biologically relevant fluids used to challenge the sensor under realistic conditions. | Blood serum/plasma, whole milk, and urine are used to test NSA and validate antifouling strategies in clinically/food-relevant contexts [1]. |
| Surface Functionalization Reagents | Chemicals used to attach antifouling layers or bioreceptors to the sensor surface. | Thiol-based linkers for gold surfaces, silanes for oxide surfaces, and cross-linkers like EDC/NHS for covalent biomolecule immobilization. |
Advanced Visualization: Impact of NSA on Biosensor Signals
Understanding the tangible impact of fouling on analytical signals is crucial for interpreting data from evaluation protocols. The following diagram illustrates how NSA manifests across different biosensor platforms.
Effective evaluation protocols for NSA are non-negotiable for the transition of biosensors from laboratory prototypes to devices capable of functioning in real-world samples like blood and milk. As demonstrated, protocols leveraging SPR, EC, and particularly coupled EC-SPR methods provide powerful, complementary data on the extent and impact of fouling [1].
The future of this field will be shaped by the high-throughput screening of novel antifouling materials, the application of molecular simulations to understand adsorption mechanisms, and the integration of machine learning-assisted evaluation to predict biosensor performance in complex environments [1]. The standardized protocols and comparative data presented in this guide provide a foundational framework that researchers can build upon to develop more robust and reliable sensing platforms.
Addressing Sensor Drift and Long-Term Stability Challenges
Sensor drift, the gradual and often unpredictable change in a sensor's response to the same stimulus over time, remains one of the most significant challenges in analytical measurement science. Within the context of characterizing nanoscale surface architectures (NSA) on sensor surfaces, understanding and mitigating drift is paramount for ensuring data reliability and reproducibility. Drift effects stem from multiple sources, including physical and chemical alterations of the sensor material (first-order drift) and uncontrollable variations in experimental conditions such as temperature or humidity (second-order drift) [57]. These phenomena lead to poor repeatability and reproducibility of readings, which is particularly problematic in long-term studies and applications requiring high precision, such as therapeutic drug monitoring and environmental sensing [57] [58].
The stability of a sensor is intrinsically linked to the properties of its surface. Characterization methods for NSA aim to link specific surface features and material compositions to the sensor's performance metrics, including its susceptibility to drift. Therefore, a critical comparison of how different sensor technologies and designs withstand the test of time is not merely a performance check but a fundamental assessment of the NSA's robustness.
Comparative Analysis of Sensor Drift Performance
The long-term stability of a sensor is a key indicator of the quality of its surface characterization and manufacturing. The following table summarizes experimental drift and stability data from published studies on different sensor types, providing a benchmark for comparison.
Table 1: Comparative Long-Term Stability Performance of Various Sensor Technologies
| Sensor Type / Product | Study Duration | Key Performance Metrics Over Time | Stability Outcome | Reference & Context |
|---|---|---|---|---|
| Metal-Oxide Gas Sensor Array (Smelldect E-nose) | 12 months | Response to diacetyl, 2-phenylethanol, and ethanol across 62 sensors. 700 time-series recordings. | Demonstrated measurable long-term drift, providing a dataset for developing compensation algorithms [57]. | [57] |
| Self-Made Integrated Three-in-One Microsensor (Velocity, Temperature, Humidity) | 744 hours (31 days) | Stable operation in HVAC ranges: 10–40°C, 60–90% RH, 1.5–5.0 m/s velocity. Overall accuracy ~±3%. | No significant signal drift observed; validated for real-world industrial HVAC environments [59]. | [59] |
| AirGradient PM2.5 Monitor (Plantower PMS5003T) | 2 years (London) & 3 months (Chennai) | RMSE, R², and slope versus BAM reference. London: Avg. slope 1.1-1.2; Chennai: Avg. slope ~0.96. | No evidence of systematic drift. Performance metrics remained within EPA targets for the duration [60]. | [60] |
| Electronic Nose with Adversarial Correction (EMAD) | 36 months (Dataset from Vergara et al.) | Classification accuracy on 6 gases over 10 batches. | Without correction, significant performance degradation. EMAD model successfully narrowed distribution gap, restoring high accuracy without needing target domain labels [61]. | [61] |
Key Insights from Comparative Data
The data reveals that drift behavior is highly technology-dependent. The metal-oxide gas sensor array explicitly showed measurable drift, a common challenge for this sensor type that necessitates advanced data correction methods [57]. In contrast, the AirGradient PM2.5 sensors exhibited remarkable stability over multiple years, suggesting that the underlying particle-counting technology and the specific NSA of the Plantower module are inherently robust or effectively compensated [60]. The MEMS-based integrated microsensor demonstrated that careful design and packaging can yield stable performance in harsh industrial environments over a continuous month of operation [59]. These differences underscore the importance of selecting sensor technology based on the required deployment duration and the acceptability of post-processing correction.
Experimental Protocols for Drift Assessment
Robust experimental design is critical for accurately quantifying sensor drift and stability. The following protocols, derived from the cited studies, provide a framework for rigorous evaluation.
Long-Term Drift Measurement in Electronic Noses
This protocol is designed to capture the long-term drift behavior of metal-oxide gas sensor arrays, crucial for developing effective drift correction algorithms [57].
- Sensor System: A commercial electronic nose (Smelldect) with an array of 62 SnO₂ sensors, operated via Kamina Observer software.
- Analytes and Concentrations: Three analytes—diacetyl, 2-phenylethanol, and ethanol (as a 5% v/v aqueous solution). Concentrations are prepared to be both substantially above and marginally above the limit of detection (LOD) to study concentration-dependent effects.
- Experimental Control:
- Pre-conditioning: Sensors are purged with dry compressed air for 24 hours before measurement cycles to minimize short-term variability.
- Stabilization: The system is connected to an empty sample bottle for ~90 minutes at the start of each day to achieve a stable sensor chamber temperature.
- Measurement Cycle: A fixed 20-minute cycle per sample, including sensor purging and sample measurement phases.
- Order of Operation: Measurements are performed daily in a fixed order as triplicates to isolate time-related drift from other variables.
- Data Collection: The dataset comprises 700 time-series recordings over 12 months. Both raw sensor data and pre-extracted features are provided to allow flexibility in developing drift compensation methods [57].
Stability Testing for Environmental Microsensors
This methodology assesses the durability and reliability of integrated sensors in real-world operating conditions, such as industrial HVAC ducts [59].
- Test Setup: The self-made sensor and two commercial velocity sensors (FS7.0.1L.195 and F660) are deployed simultaneously in the same industrial HVAC environment for direct comparison.
- Environmental Ranges: The sensors are evaluated over specified operational ranges: 10–40 °C for temperature, 60–90% RH for humidity, and 1.5–5.0 m/s for airflow velocity.
- Duration: The long-term test is conducted continuously for 744 hours.
- Data Acquisition & Comparison: A wireless MQTT/Node-RED architecture is used for real-time, continuous data logging. The output of the self-made sensor is directly compared against the commercial sensors and the known system parameters to assess accuracy and the absence of drift over the extended period [59].
Field-Based Drift Assessment for Air Quality Sensors
This protocol uses co-location with reference instruments to evaluate the long-term stability and potential drift of low-cost PM2.5 sensors in the field [60].
- Site Selection: Multiple locations with varying pollution levels (e.g., low/medium in London, high in Chennai) are chosen to assess performance across different concentration ranges.
- Reference Instrument: The sensor units (e.g., AirGradient monitors) are co-located with a reference-grade instrument, specifically a Beta Attenuation Monitor (BAM).
- Data Processing and Metrics: Sensor data is corrected according to US EPA guidelines and aggregated to a 24-hour resolution. Key performance metrics are calculated periodically (e.g., monthly or weekly):
- Root Mean Square Error (RMSE): Measures average sensor error.
- Coefficient of Determination (R²): Quantifies how well the sensor captures trends from the reference.
- Slope: Represents the sensor's sensitivity relative to the reference.
- Coefficient of Variation (CV): Assesses precision between multiple co-located sensors.
- Drift Analysis: The temporal trends of these metrics (e.g., a consistent increase or decrease in the slope) are analyzed over the study period (e.g., 2 years) to identify any systematic drift [60].
Advanced Drift Correction Algorithms and Mechanisms
When hardware design and calibration alone cannot eliminate drift, algorithmic correction methods are essential. A leading approach involves treating drift as a domain adaptation problem.
The EMAD: An Adversarial Network for Drift Correction
The Entropy Minimization model based on Adversarial Networks (EMAD) is designed to correct for sensor drift without requiring labeled data from the drifted sensors (the "target domain") [61].
- Core Mechanism: The model consists of a generator (acting as a feature extractor) and a classifier. The key innovation is reusing the classifier as a discriminator in an adversarial training setup. The generator aims to produce features that are indistinguishable between the original (source) and drifted (target) data, thereby narrowing the distribution gap caused by drift. Simultaneously, the classifier is trained to correctly label the source domain data.
- Preserving Discriminability: To prevent the "mode collapse" where all data maps to a single point, EMAD incorporates conditional entropy minimization and feature norm loss. This ensures that while features from both domains are aligned, they do not overlap in a way that destroys classification boundaries [61].
- Workflow: The process involves feeding both pristine source data and unlabeled, drifted target data into the network. Through adversarial training, the generator learns to create a domain-invariant feature space. The final classifier can then accurately predict labels for the drifted data based on this invariant space.
The following diagram illustrates the architectural workflow and the adversarial training process of the EMAD model.
Diagram 1: EMAD Adversarial Correction Workflow. The model uses adversarial training to force the generator to produce features that are indistinguishable between source and target domains, allowing the classifier to work accurately on drifted data.
The Scientist's Toolkit: Essential Reagents and Materials
The following table details key reagents, materials, and software tools used in the experimental protocols for sensor drift characterization and correction, as discussed in this guide.
Table 2: Key Research Reagent Solutions for Sensor Drift Characterization
| Item Name | Function / Application | Specific Example from Context |
|---|---|---|
| Diacetyl & 2-Phenylethanol | Model volatile organic compounds (VOCs) for gas sensor drift testing. Provide different chemical moieties and vapor pressures. | Used as analytes in the 12-month metal-oxide sensor array drift study [57]. |
| Ethanol (5% v/v Aqueous Solution) | Acts as both a diluent for other analytes and a sample/blank control in gas sensor measurements. | Served as a control and diluent in the long-term E-nose dataset [57]. |
| Chitosan-Sodium Chloride Composite | Sensing material for high-performance capacitive humidity sensors. | Used in micro humidity sensors to achieve a 72-fold increase in capacitive response [59]. |
| Tyrosinase Enzyme | Biorecognition element for specific electrochemical detection of Levodopa (L-Dopa). | Immobilized on a screen-printed carbon electrode in a wearable sensor for Parkinson's drug monitoring [58]. |
| Kamina Observer Software | Controls commercial electronic nose systems for automated measurement protocols. | Used to operate the Smelldect E-nose and manage its multi-phase measurement cycles [57]. |
| Node-RED with MQTT Protocol | Open-source platform for wiring IoT devices and enabling real-time data acquisition. | Implemented in the HVAC microsensor study for wireless, real-time data flow from factory ducts [59]. |
Addressing sensor drift is a multi-faceted challenge requiring a combination of robust sensor design, rigorous long-term testing, and sophisticated algorithmic correction. The comparative data shows that while some sensor technologies exhibit inherent stability, others are prone to significant drift that must be compensated. The experimental protocols for drift assessment provide a blueprint for generating reliable, quantitative data on sensor performance over time. Furthermore, advanced machine learning techniques, such as the EMAD adversarial network, offer powerful, data-driven solutions for mitigating drift effects, ensuring that sensors remain accurate and reliable throughout their operational lifespan. For researchers characterizing nanoscale surface architectures, integrating these long-term stability assessments and correction strategies is essential for developing next-generation, drift-resilient sensors.
Benchmarking NSA Characterization Methods: Performance Metrics and Validation Frameworks
The performance of biosensors is critically dependent on the effective suppression of non-specific adsorption (NSA), a phenomenon where molecules indiscriminately bind to sensing surfaces, leading to elevated background signals, false positives, and reduced reliability [18]. The characterization of NSA on sensor surfaces presents a significant challenge in the development of sensitive diagnostic tools, particularly for detecting low-abundance protein biomarkers in clinical settings [18] [62]. This guide provides a comparative analysis of techniques used to quantify and mitigate NSA, focusing on the critical parameters of sensitivity, throughput, and cost for researchers and drug development professionals working within the broader context of sensor characterization methodology.
Non-specific adsorption, also known as biofouling, occurs when proteins or other biomolecules physisorb to a sensor's surface via hydrophobic forces, ionic interactions, van der Waals forces, or hydrogen bonding [18]. This non-specific binding can result in four distinct interference types: (1) molecules adsorbed on vacant spaces, (2) molecules adsorbed on non-immunological sites, (3) molecules adsorbed on immunological sites while still allowing antigen access, and (4) molecules adsorbed on immunological sites that block antigen binding [18]. The consequences manifest as compromised sensitivity, specificity, dynamic range, and reproducibility of biosensing platforms [18].
The balance between specific sensing and NSA reduction represents a fundamental challenge in biosensor design, particularly as sensors move toward miniaturization where the size of probe molecules and analytes becomes comparable to the sensitive area of the sensor itself [18]. This has driven the development of numerous characterization methods to quantify NSA and evaluate the efficacy of mitigation strategies.
Comparative Analysis of NSA Characterization Techniques
Various analytical techniques are employed to characterize NSA, each with distinct operating principles, sensitivity limits, and application suitability. The table below summarizes the key techniques used in NSA research.
Table 1: Comparison of Techniques for Characterizing NSA on Sensor Surfaces
| Technique | Principle of Operation | Sensitivity (Detection Limit) | Throughput | Relative Cost | Key Applications in NSA Research |
|---|---|---|---|---|---|
| Fluorescence Microscopy [62] | Detection of fluorescently labeled proteins (e.g., FITC-BSA) | High (single-protein detection possible with advanced setups) | Moderate | Moderate | Quantitative assessment of protein adsorption on modified surfaces (e.g., PMMA) |
| Surface Plasmon Resonance (SPR) [18] | Measures refractive index changes near a metal surface | ~0.3 nM for heavy metals [63] | Low to Moderate | High | Real-time monitoring of binding events; evaluation of antifouling surfaces |
| Ellipsometry [18] | Measures polarization change of reflected light | Nanometric thickness changes [18] | Moderate | High | Label-free study of protein adsorption; can differentiate specific from non-specific binding in some cases |
| Atomic Force Microscopy (AFM) [62] | Physical probing of surface topography with a nanoscale tip | Sub-nanometer vertical resolution | Low | High | Surface microstructure characterization and nanoscale roughness measurement |
| Attenuated Total Reflection Fourier Transform Infrared (ATR-FTIR) [18] [62] | Detects molecular vibrational signatures via evanescent wave | Single protein layer detection [18] | Moderate | High | Chemical composition analysis of modified surfaces; identification of adsorbed species |
| Ion-Current-Rectifying Nanopore Sensing [64] | Measures modulation in current-voltage curve due to surface charge changes | Potentially ultrasensitive; optimized for specific analyte detection [64] | Low (device-to-device variation) | Low to Moderate (depending on fabrication) | Sensitive to surface charge modulation from analyte binding; limited by reproducibility |
Key Insights from Technique Comparison
Sensitivity Considerations: Nanoscale sensors, including nanopores, can achieve ultra-high sensitivity due to properties such as large relative surface area and confined responsive regions [64]. However, their sensitivity is profoundly affected by both geometric parameters (e.g., pore size, cone angle) and operating conditions (e.g., electrolyte concentration) [64]. For optical techniques, emerging methods like scattering lens microscopy can improve resolution by fivefold, enabling subcellular imaging without labeling [65].
Throughput and Reproducibility: A significant trade-off exists between sensitivity and throughput. While techniques like fluorescence microscopy offer reasonable throughput, nanoscale sensing systems often suffer from low device-to-device reproducibility, requiring multiple repetitions for statistical significance [64]. Characterization techniques with wider fields of view (hundreds of micrometers) help address the scale range problem in studying nanostructures within larger biological contexts [65].
Cost Implications: Techniques requiring sophisticated instrumentation like SPR, ellipsometry, and AFM entail high capital investment. In contrast, fluorescence microscopy setups offer a more moderate-cost option with versatile application. Fabrication costs for nanoscale sensors can vary significantly based on the need for cleanroom facilities [64].
Experimental Protocols for NSA Quantification
Fluorescence-Based NSA Assessment on Modified Polymer Surfaces
This protocol, adapted from research on poly(methyl methacrylate) (PMMA) surfaces, details the quantification of protein adsorption using fluorescence labeling [62].
Surface Modification: PMMA substrates are modified using techniques such as bovine serum albumin (BSA) coating, polyethylene glycol (PEG) grafting, or plasma cleaning. For BSA coating, surfaces are incubated with BSA solution (e.g., 1% w/v) for a defined period (e.g., 1 hour) followed by washing to remove unbound protein [62].
Protein Labeling: Bovine serum albumin is labeled with fluorescein isothiocyanate (FITC) following standard conjugation protocols. Unbound FITC is removed through dialysis or gel filtration [62].
Adsorption Experiment: Modified surfaces are incubated with FITC-BSA solution under controlled conditions (concentration, time, temperature, pH 7.4). A typical concentration range is 0.1-1.0 mg/mL with incubation times from 30 minutes to 2 hours [62].
Quantification: After washing to remove non-adsorbed protein, fluorescence intensity is measured using fluorescence microscopy or a plate reader. The percentage of anti-protein adsorption effect is calculated as:
[1 - (Fluorescence of test surface / Fluorescence of control surface)] × 100%[62]. Studies report BSA coating achieving over 87.6% anti-adsorption effect, with plasma cleaning at 86.1% [62].
Finite Element Analysis for Nanoscale Sensor Optimization
For nanoscale sensors like ion-current-rectifying nanopores, computational optimization is crucial before experimental validation due to low throughput and high prototyping costs [64].
Model Construction: Create a accurate geometric model of the sensor (e.g., conical nanopore) using simulation software. Define relevant geometric parameters (pore radius, cone angle) and operating conditions (electrolyte concentration, applied voltage) [64].
Parameter Sampling: Sample the input parameter space using distributions that reflect real-world random errors in fabrication and measurement (e.g., normal distribution of pore radii) [64].
Sensitivity Analysis: Employ Morris method screening to identify influential parameters, followed by quantitative Sobol analysis to determine how much of the output variance each input parameter explains [64].
Optimization: Calculate Jeffreys' divergence to quantify dissimilarity between output distributions with and without analyte present. Optimize geometric and operating parameters to maximize this dissimilarity, indicating higher sensitivity [64]. Studies indicate highest sensitivity for larger pores operated at low electrolyte concentrations [64].
The following DOT script describes the workflow for the computational approach to nanoscale sensor optimization:
Diagram 1: Computational Optimization Workflow for Nanoscale Sensors. This workflow illustrates the process for optimizing sensor sensitivity using finite element analysis before experimental validation.
Visualization of NSA Reduction and Characterization Strategies
Understanding the relationships between NSA reduction strategies, characterization methods, and sensor performance parameters is essential for selecting appropriate techniques. The following diagram maps these connections to guide research design.
The following DOT script describes the logical relationships between NSA reduction methods, characterization techniques, and performance metrics:
Diagram 2: NSA Reduction and Characterization Framework. This diagram illustrates the relationships between different NSA reduction strategies, characterization techniques, and key performance evaluation metrics.
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 2: Key Research Reagent Solutions for NSA Research
| Reagent/Material | Function in NSA Research | Example Application |
|---|---|---|
| Bovine Serum Albumin (BSA) [18] [62] | Blocking agent that passively adsorbs to surfaces to reduce NSA | Coating PMMA microfluidic channels; achieving >87.6% reduction in protein adsorption [62] |
| Polyethylene Glycol (PEG) [18] [62] | Polymer grafting creates hydrophilic, non-charged boundary layer to prevent protein adsorption | Grafting on sensor surfaces to minimize intermolecular forces with adsorbing molecules [18] [62] |
| Fluorescein Isothiocyanate (FITC) [62] | Fluorescent label for proteins to enable quantification of adsorption | Labeling BSA for fluorescence-based NSA assessment on modified surfaces [62] |
| Self-Assembled Monolayers (SAMs) [18] | Linker molecules to improve surface immobilization and patterning of functional molecules | Creating controlled patterning of functional molecules on biosensor surfaces [18] |
| Casein and Milk Proteins [18] | Protein-based blocking agents that passively coat surfaces | Reducing NSA in immunoassays like ELISA and Western blotting [18] |
| Plasma Treatment Systems [62] | Surface activation and cleaning method to modify surface properties | PMMA surface treatment achieving 86.1% anti-protein adsorption effect [62] |
The characterization of non-specific adsorption on sensor surfaces requires careful selection of techniques balanced against sensitivity requirements, throughput needs, and cost constraints. Fluorescence-based methods offer a practical balance for many applications, while specialized techniques like SPR and ellipsometry provide detailed, label-free analysis at higher investment. For emerging nanoscale sensors, computational optimization through finite element analysis has become an essential precursor to experimental validation, addressing challenges of reproducibility and fabrication cost. The ongoing development of both super-resolution and nanodetection optical techniques continues to push the boundaries of what is measurable in NSA characterization, enabling researchers to develop more reliable biosensors for diagnostic and drug development applications.
Establishing Standardized Metrics for Antifouling Efficacy
The accumulation of biological fouling (biofouling) on sensor surfaces presents a significant challenge in marine research, drug development, and environmental monitoring. Biofouling can impair sensor accuracy, reduce operational lifespan, and compromise data integrity. A critical barrier to developing effective antifouling (AF) strategies has been the lack of standardized, quantitative metrics to evaluate coating efficacy across different laboratories and environments. This guide objectively compares current antifouling evaluation methods, supported by experimental data, to empower researchers in selecting and developing robust characterization protocols for sensor surface protection.
Comparative Analysis of Antifouling Evaluation Methods
The evaluation of antifouling efficacy spans laboratory bioassays and field trials, each offering distinct advantages and limitations. The table below summarizes the key characteristics of current and emerging methods.
Table 1: Standardized Metrics and Methods for Antifouling Efficacy Evaluation
| Method Name | Test Organism/System | Key Measured Parameters | Throughput | Real-World Correlation | Primary Application | Standardization Status |
|---|---|---|---|---|---|---|
| Single Thread Tensile Adhesion Test (STAT) [66] [67] | Mussel (Mytilus sp.) | Tensile adhesion force of individual byssal threads (kPa) | Medium | High (for adhesion) | High-performance foul-release coatings | Emerging protocol (2025) |
| Flow-Through Laboratory Bioassay [68] | Mussel (M. galloprovincialis) | Number of byssus threads produced; Biocide leaching rate | Low | Medium | Biocide-based paint screening | Laboratory standard |
| ISO 21716 Series [69] | Barnacles, Mussels, Algae (Ectocarpus sp.) | Settlement inhibition; Growth inhibition | Medium to High | Under validation | Broad-spectrum AF paint screening | International Standard (2025) |
| Field Immersion Trials [70] [71] | Multi-species fouling community | Fouling coverage (%); Biovolume; Species identification | Very Low | Very High | Final product validation | Industry standard, site-dependent |
| Brown Algae Bioassay (ISO 21716-4) [69] | Brown Algae (Ectocarpus sp.) | Growth inhibition | High | To be determined | Early-stage screening of AF paints | Newly published standard (2025) |
Detailed Experimental Protocols and Data
Tensile Adhesion Method for Foul-Release Coatings
The Single Thread Tensile Adhesion Test (STAT) is an advanced method designed to quantitatively measure the adhesion strength of mussel byssus threads, providing high resolution for comparing high-performance foul-release coatings [66] [67].
Table 2: Experimental Data from Mussel Adhesion Tests
| Coating Type | Test Method | Average Adhesion Force | Resolution | Key Finding |
|---|---|---|---|---|
| Commercial Foul-Release 1 | STAT [67] | Low (Precise value in kPa) | High | Effectively discriminates between high-performance coatings. |
| Commercial Foul-Release 2 | STAT [67] | Significantly Higher | High | STAT detected differences unseen by other methods. |
| Biocide-based (0% Cu₂O) | Flow-Through Bioassay [68] | High (Numerous byssus threads) | Low | Byssus thread count inversely related to biocide content. |
| Biocide-based (40% Cu₂O) | Flow-Through Bioassay [68] | Low (Few byssus threads) | Low | Confirmed high efficacy of high biocide content. |
Protocol: Single Thread Tensile Adhesion Test (STAT) [66] [67]
- Surface Preparation: Coat substrates with the AF coatings to be tested and condition in filtered seawater for at least 24 hours.
- Mussel Acclimation: Individually house mussels in a flow-through seawater system.
- Thread Attachment: Place a single mussel on the coated surface and allow it to attach a byssal thread. This process can be guided towards a specific area.
- Force Measurement: Using a microtensile tester, individually grip the byssal plaque and the thread. Apply a tensile force perpendicular to the coating surface at a constant displacement rate until failure occurs.
- Data Analysis: Record the maximum force required for detachment. A minimum of 30 replicates per coating is recommended for statistical power.
Laboratory Bioassay for Biocide-Based Paints
This protocol uses mussel behavior to evaluate the efficacy of biocide-releasing paints under controlled, dynamic conditions [68].
Protocol: Flow-Through Mussel Bioassay [68]
- Paint Aging: Dynamically age painted test plates (e.g., PVC or steel) in a flow-through seawater system. A rotating cylinder apparatus simulates vessel movement, aging surfaces at a speed of ~10 knots for 45 days.
- Biofilm Removal: Gently wipe the aged plate surfaces to remove microbial biofilm, which can influence mussel settlement.
- Experimental Setup: Place one aged test plate in a glass tank with continuous flow of filtered seawater (e.g., ~14 mL/min, achieving ~10 water exchanges per day).
- Mussel Introduction: Securely paste five young mussels (Mytilus galloprovincialis) onto the coated surface of each test plate.
- Response Measurement: After a set period (e.g., 24-48 hours), count the number of byssus threads secreted by each mussel. A lower count indicates higher coating efficacy.
- Biocide Leaching Analysis: Measure the concentration of biocides (e.g., Cu₂O) in the test water using ICP-MS after solid-phase extraction.
Field Immersion Trials for Holistic Validation
Field trials provide the most direct measure of a coating's performance under realistic, multi-species fouling pressure [70] [71].
Protocol: Static Field Immersion [71]
- Panel Preparation: Incorporate the AF agent (e.g., a natural extract like Bacillus licheniformis MCE) into a paint matrix at desired concentrations (e.g., 2% and 5% w/w). Apply the paint to test panels.
- Site Deployment: Immerse the treated and control panels in a natural marine environment (e.g., a harbor or gulf) for an extended period, typically 90-180 days.
- Data Collection: Periodically document fouling coverage (%) and biomass on the panels. Identify the dominant fouling species.
- Efficacy Analysis: Compare fouling coverage and community composition between treated and control panels. For example, a 5% MCE-treated panel showed a 30% reduction in fouling coverage compared to the control after 180 days [71].
Signaling Pathways and Experimental Workflows
The following diagrams illustrate the logical relationships and experimental workflows for key antifouling evaluation methods.
Antifouling Evaluation Pathway Logic
Single Thread Tensile Test Workflow
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 3: Key Reagents and Materials for Antifouling Efficacy Research
| Item | Function/Description | Example Use Case |
|---|---|---|
| Self-Polishing Copolymer Resin | Matrix polymer that controls the gradual release of biocides from the paint film [68]. | Base for biocide-based AF paints in dynamic aging tests [68]. |
| Cuprous Oxide (Cu₂O) | Inorganic biocide that prevents fouling organism settlement through toxicity [68]. | Primary active ingredient in formulated test paints; used to establish dose-response curves [68]. |
| Mussel (Mytilus galloprovincialis) | Model macro-fouling organism; its byssus thread production is a key metric for adhesion [68] [67]. | Used in STAT, flow-through bioassays, and field trials to assess coating performance [68] [67]. |
| Brown Algae (Ectocarpus sp.) | Model micro-fouling organism for standardized growth inhibition tests [69]. | Screening antifouling paints for anti-algal properties according to ISO 21716-4 [69]. |
| Methanol Cell Extract (MCE) | Natural product extract with anti-biofilm and anti-settlement properties [71]. | Incorporated into paints (e.g., 5% w/w) as an environmentally friendly antifouling agent for field testing [71]. |
| Dynamic Aging Apparatus | System to simulate hydrodynamic conditions and polish paint surfaces under controlled lab settings [68]. | Aging test plates coated with AF paints before bioassay to mimic real-world conditioning [68]. |
Non-specific adsorption (NSA) is a fundamental challenge that impedes the widespread adoption and reliability of biosensors in clinical, environmental, and food safety applications [1]. NSA refers to the undesirable accumulation of non-target molecules (e.g., proteins, lipids, cells) on the biosensing interface, which compromises analytical performance by causing false positives, elevated background signals, reduced sensitivity, and impaired reproducibility [1] [18]. The detrimental impact of NSA intensifies when analyzing complex biological samples such as blood, serum, and milk, where high concentrations of interfering species coexist with the target analyte [1].
Addressing NSA requires a detailed understanding of its mechanisms and the deployment of effective mitigation strategies tailored to specific transducer principles. This guide provides a systematic comparison of NSA analysis and mitigation in two prominent biosensor platforms: electrochemical (EC) and surface plasmon resonance (SPR) biosensors. A emerging powerful tool in this field is the use of coupled electrochemical-surface plasmon resonance (EC-SPR) systems, which provide complementary data to better understand and quantify interfacial fouling [1] [72]. By examining experimental case studies and performance data, this review aims to equip researchers with the knowledge to select appropriate characterization methods and antifouling strategies for their specific biosensing applications.
Fundamental Mechanisms of NSA and Its Biosensor Impacts
Origins and Driving Forces of NSA
The accumulation of non-target sample components on biosensor surfaces occurs primarily through physisorption, driven by a combination of electrostatic interactions, hydrophobic forces, hydrogen bonding, and van der Waals forces [1] [18]. These interactions are influenced by the physicochemical properties of both the sensor surface and the interfering molecules present in the sample matrix.
Consequences for Biosensor Signal Integrity
The presence of NSA manifests differently depending on the biosensing mechanism, but consistently degrades key performance parameters:
- In electrochemical biosensors, fouling layers can passivate the electrode surface, hinder electron transfer kinetics, increase impedance, and cause signal drift over time [1]. For structure-switching aptamer-based sensors, NSA can restrict the conformational freedom needed for target recognition [1].
- In SPR biosensors, non-specifically adsorbed molecules contribute to reflectivity changes that are optically indistinguishable from specific binding events, leading to overestimation of analyte concentration [1]. This directly affects binding kinetics analysis and quantification accuracy.
The diagram below illustrates the multifaceted impact of NSA across different biosensor platforms:
Figure 1: NSA Impacts on Different Biosensor Platforms
Analytical Methods for NSA Quantification
Electrochemical NSA Assessment Techniques
Electrochemical methods leverage changes in electrical properties to monitor fouling in real-time:
- Electrochemical impedance spectroscopy (EIS) tracks increases in charge-transfer resistance resulting from insulting fouling layers.
- Chronoamperometry monitors decay in Faradaic current as non-conductive species adsorb onto the electrode surface.
- Cyclic voltammetry detects changes in peak current and peak separation, indicating hindered electron transfer kinetics.
SPR NSA Assessment Techniques
SPR platforms detect NSA through changes in optical properties at the sensor surface:
- Angle shift measurements track changes in resonance angle due to adsorption of non-target biomolecules.
- Reflectivity imaging enables spatial mapping of fouling across the sensor surface.
- Kinetic analysis distinguishes specific binding (typically slower, concentration-dependent) from rapid, non-specific adsorption.
Advantages of Coupled EC-SPR Systems
Integrated EC-SPR platforms provide complementary NSA assessment by simultaneously monitoring both electrochemical (e.g., current, impedance) and optical (reflectivity) parameters [1] [72]. This multi-dimensional approach offers several advantages:
- Cross-validation of NSA phenomena through orthogonal detection methods
- Enhanced quantification of fouling layers by correlating optical thickness with electrochemical passivation
- Comprehensive interfacial characterization by combining SPR's mass sensitivity with EC's charge sensitivity
Comparative Analysis: NSA in Electrochemical vs. SPR Biosensors
Table 1: Direct Comparison of NSA Characteristics in Electrochemical and SPR Biosensors
| Parameter | Electrochemical Biosensors | SPR Biosensors |
|---|---|---|
| Primary NSA Manifestation | Passivation layer increasing charge-transfer resistance [1] | Bulk refractive index change masking specific binding [1] |
| Key Impacted Metrics | Sensitivity, electron transfer rate, signal stability [1] | Binding affinity quantification, detection limit, specificity [1] [73] |
| NSA Detection Methods | EIS, voltammetry, chronoamperometry [1] | Resonance angle shift, reflectivity imaging [1] |
| Typical LOD Degradation | Can increase LOD by 1-2 orders of magnitude in complex samples | Can increase LOD from fM to pM range without mitigation [73] |
| Optimal NSA Assessment | Real-time monitoring of current/ impedance decay [1] | Real-time angle shift tracking with reference channels [73] |
Table 2: NSA Mitigation Strategies in Electrochemical vs. SPR Biosensors
| Strategy Type | Electrochemical Approaches | SPR Approaches |
|---|---|---|
| Antifouling Coatings | Conducting polymers, peptides, hybrid materials, cross-linked protein films [1] | Hydrogels, poly(ethylene glycol), zwitterionic materials [1] [18] |
| Surface Chemistry | Self-assembled monolayers with terminal oligo(ethylene glycol) [18] | Carboxymethyl dextran hydrogels for biomolecule immobilization [18] |
| Active Removal Methods | Electrochemical cleaning (potential cycling) [18] | Microfluidic shear forces, electromechanical transducers [18] |
| Sample Treatment | Dilution, centrifugation, addition of blocking agents [1] | Sample dilution, buffer optimization, surfactant addition [1] |
| Signal Correction | Background subtraction using control electrodes [1] | Reference channel subtraction [73] |
Case Studies in NSA Analysis
Electrochemical Case Study: Aptamer-Based Biosensor
A recent investigation examined NSA impacts on electrochemical aptamer-based (E-AB) biosensors in undiluted serum [1]. Researchers observed significant signal degradation over time, manifested as both signal drift and reduced target-induced signal change [1].
Experimental Protocol:
- Sensor Fabrication: Gold electrodes were functionalized with methylene-blue-modified DNA aptamers specific to a model protein target.
- NSA Assessment: EIS and square-wave voltammetry were performed before and after exposure to 100% serum.
- Mitigation Strategy: Co-immobilization of zwitterionic peptides around the aptamer layer.
- Performance Metrics: Signal retention (%) and LOD shift were quantified.
Results: Unprotected sensors exhibited >80% signal loss within 30 minutes, while peptide-modified sensors maintained >70% initial signal after 2 hours. The LOD was preserved within one order of magnitude for protected sensors versus three orders of magnitude degradation for unprotected interfaces.
SPR Case Study: DNA Hybridization Sensor
A 2021 study developed a self-referencing SPR biosensor with ultralow limit-of-detection for DNA hybridization, specifically addressing NSA challenges [73].
Experimental Protocol:
- Sensor Design: Implemented long-wavelength excitation (LW-SPR) achieving greater decay length (~1000 nm) and self-referencing channel.
- Surface Functionalization: DNA probes immobilized onto a mixed poly(ethylene glycol) monolayer.
- NSA Assessment: Compared signal response in specific vs. reference channels.
- Signal Amplification: Employed graphene oxide-assisted Au nanoparticle conjugates.
Results: The self-referencing approach effectively discriminated NSA from specific DNA binding, achieving an LOD of 0.2 fM for target DNA even in complex samples. The reference channel corrected for bulk refractive index changes and temperature fluctuations that often obscure specific signals [73].
Coupled EC-SPR Case Study: Protein Binding Analysis
A coupled EC-SPR platform was used to investigate NSA during protein-drug interaction studies in serum-like conditions [1].
Experimental Protocol:
- Platform Configuration: SPR gold chip served as working electrode in a three-electrode electrochemical cell.
- Surface Modification: Coated with a conductive antifouling polymer combining PEG-like properties with aniline conductivity.
- Simultaneous Monitoring: Tracked both SPR angle shift and electrochemical impedance during protein exposure.
- Data Correlation: Cross-referenced optical thickness changes with charge transfer resistance.
Results: The coupled system provided complementary NSA quantification, revealing that approximately 60% of the SPR signal shift in complex samples originated from non-specific adsorption rather than target binding. This enabled more accurate kinetic constant determination for the specific interaction [1].
Research Reagent Solutions for NSA Reduction
Table 3: Essential Reagents for NSA Mitigation Research
| Reagent Category | Specific Examples | Function & Mechanism |
|---|---|---|
| Blocking Proteins | BSA, casein, milk proteins [18] | Adsorb to surface空白s, reducing non-specific protein binding |
| Chemical Linkers | 11-mercaptoundecanoic acid (MUA), HS-PEG-OH [74] | Form self-assembled monolayers that resist protein adsorption |
| Zwitterionic Materials | Poly(carboxybetaine), poly(sulfobetaine) [18] | Create superhydrophilic surfaces through strong water binding |
| Hydrogels | Carboxymethyl dextran, polyacrylamide [18] | Form hydrated physical barriers against foulant approach |
| Conductive Polymers | Poly(aniline), poly(3,4-ethylenedioxythiophene) with antifouling side chains [1] | Combine electron transfer capability with fouling resistance |
| Nanoparticles | Gold nanoparticles, graphene oxide [73] | Signal amplification tags that enhance sensitivity and specificity |
Experimental Design for NSA Analysis
The diagram below outlines a generalized workflow for conducting NSA analysis in biosensor development:
Figure 2: Experimental Workflow for NSA Analysis
The evolving landscape of NSA research points toward several promising directions. Advanced materials including new peptide-based coatings, hybrid polymers, and stimuli-responsive layers offer increasingly sophisticated antifouling capabilities while maintaining biosensor functionality [1]. Multimodal sensing platforms, particularly coupled EC-SPR systems, provide more comprehensive NSA quantification by combining orthogonal detection principles [1] [72]. Emerging computational approaches employing molecular simulations and machine learning are accelerating the discovery and optimization of antifouling interfaces [1].
For researchers selecting between electrochemical and SPR platforms for NSA-sensitive applications, the decision framework should consider:
Electrochemical biosensors offer advantages in point-of-care settings due to their miniaturization potential, lower cost, and compatibility with complex samples when appropriate antifouling strategies are implemented.
SPR biosensors provide superior capabilities for detailed binding kinetics studies and benefit from well-established reference channel methodologies for NSA correction.
Coupled EC-SPR systems represent the most powerful approach for fundamental NSA characterization and development of novel antifouling strategies, despite their greater complexity.
The systematic analysis of NSA remains essential for advancing biosensor technology toward real-world applications in clinical diagnostics, environmental monitoring, and food safety. As antifouling methodologies continue to evolve, the integration of material science, surface chemistry, and multi-modal detection will further bridge the gap between laboratory demonstrations and field-deployable biosensing solutions.
The characterization of sensor surfaces, particularly for quantifying non-steroidal anti-inflammatory drugs (NSAIDs), is undergoing a transformative shift driven by technological advancements in high-throughput screening (HTS) and machine learning (ML). Traditional analytical methods for NSAID detection and quantification—including high-performance liquid chromatography (HPLC), gas chromatography (GC), and mass spectrometry—offer high sensitivity and selectivity but are often constrained by high operational costs, lengthy analysis times, and requirements for sophisticated laboratory infrastructure and specialized personnel [75] [76]. These limitations become particularly pronounced in pharmaceutical and clinical settings where rapid, cost-effective, and large-scale analysis is essential for drug discovery, environmental monitoring, and therapeutic drug monitoring.
In response to these challenges, the integration of HTS methodologies with ML algorithms has created new paradigms for accelerating sensor development and characterization. HTS enables the rapid experimental testing of thousands of chemical compounds or sensor configurations, while ML models excel at identifying complex, non-linear patterns within the large, multidimensional datasets generated by these screens [77]. This powerful combination is proving particularly valuable for optimizing sensor materials and configurations for NSAID detection, where factors such as sensitivity, selectivity, and operational stability must be balanced across diverse and complex sample matrices including biological fluids and environmental waters [78] [76]. This guide provides a comprehensive comparison of these emerging approaches against traditional methods, supported by experimental data and detailed protocols, to inform researchers and drug development professionals about the current state and future trajectory of sensor characterization technologies.
Comparative Analysis of Screening and Design Methodologies
The table below provides a systematic comparison of the core methodologies used in NSAID sensor research, highlighting the evolution from traditional analytical techniques to modern HTS and ML-assisted approaches.
Table 1: Comparison of Methodologies for NSAID Sensor Research and Characterization
| Methodology | Key Principles | Typical Applications in NSAID Research | Throughput | Key Advantages | Major Limitations |
|---|---|---|---|---|---|
| Traditional Analytical Techniques [75] [76] | Physical separation and quantification based on chemical properties (e.g., polarity, mass). | Reference method for validation; quantification of NSAIDs in pharmaceuticals and biological samples. | Low (Manual, sequential samples) | High sensitivity and reproducibility; well-established protocols. | High cost, lengthy analysis, complex equipment, requires specialized operators. |
| High-Throughput Screening (HTS) [79] [80] | Automated, parallel testing of thousands of samples using robotics and microplate formats. | Rapid assessment of electrode materials, binding agents, or sensor formulations for NSAID detection. | Very High (Thousands of data points per day) | Accelerates empirical discovery; identifies lead candidates from vast libraries. | High initial setup cost; generates complex datasets that require sophisticated analysis. |
| Quantitative HTS (qHTS) [79] | HTS performed across a range of concentrations to generate concentration-response data. | Profiling sensor response dynamics and cross-reactivity; establishing limits of detection (LOD). | High | Generates rich, multi-parametric data for each test condition; improves reliability. | Data analysis is computationally intensive; requires careful statistical handling to avoid false positives/negatives. |
| Machine Learning (ML)-Assisted Design [81] [77] | Algorithms learn from existing data to predict properties and optimize designs without explicit programming. | Predicting sensor-analyte interactions; virtual screening of materials/nanostructures; optimizing sensor performance. | Extremely High (Once trained) | Reduces experimental trial-and-error; uncovers non-intuitive design rules; accelerates optimization. | Requires large, high-quality datasets; "black box" interpretation challenges; risk of overfitting. |
| ML-Accelerated HTS [81] [82] | ML guides HTS experimental design and analyzes HTS-generated data to extract meaningful patterns. | Integrated workflow for the rapid discovery and optimization of novel sensor materials and morphologies. | High (Intelligent, focused screening) | Maximizes information yield from experiments; creates a virtuous cycle of improvement. | Highest complexity; requires interdisciplinary expertise in both experimental and data sciences. |
Experimental Protocols for High-Throughput and ML-Driven Workflows
Protocol for qHTS of Nanomaterial-Based Sensor Libraries
This protocol is adapted from recent work on screening nanoparticle morphologies and is tailored for evaluating libraries of sensor materials for NSAID detection [81] [79].
- Library Synthesis and Preparation: Generate a diverse library of candidate sensor materials (e.g., carbon nanotubes functionalized with different polymers, metal nanoparticles of varying shapes and sizes, or molecularly imprinted polymers with different templates). This can be achieved using automated synthesizers or combinatorial chemistry techniques.
- Assay Miniaturization and Automation: Dispense the sensor material suspensions into 1536-well microplates using robotic liquid handlers. Each well represents a unique sensor formulation.
- Concentration-Response Testing: For each sensor material, introduce a series of concentrations of the target NSAID (e.g., diclofenac or ibuprofen) across different wells. A typical qHTS may test 10-15 concentrations to establish a full response profile [79].
- Signal Acquisition: Use high-sensitivity detectors to measure the electrochemical (e.g., current, potential change) or optical (e.g., fluorescence, absorbance) signal from each well simultaneously or in rapid sequence.
- Data Preprocessing: Apply robust data correction algorithms to remove systematic noise and bias, such as plate-wide row or column effects, using methods like trimmed-mean polish [80].
- Dose-Response Modeling: Fit the corrected data to a nonlinear model, such as the Hill equation, to extract key performance parameters for each sensor material, including:
- AC~50~: The concentration eliciting half-maximal signal, indicating apparent affinity.
- E~max~: The maximal response, indicating efficacy.
- Hill Slope (h): The steepness of the response curve. The reliability of these parameter estimates is highly dependent on the concentration range covering at least one of the asymptotes of the response curve [79].
- Hit Identification: Use statistical inference models, such as the RVM t-test, to benchmark the performance of each sensor material against negative controls, declaring materials with significant and desirable response profiles as "hits" for further development [80].
Protocol for Machine Learning-Assisted Sensor Optimization
This protocol outlines a ligand-based drug design approach, repurposed for predicting and optimizing sensor-analyte interactions for NSAIDs [77].
- Feature Extraction (for Traditional ML): For each candidate sensor material or molecular receptor, calculate a set of quantitative descriptors. These may include:
- Chemical Descriptors: Molecular weight, topological surface area, logP (hydrophobicity), number of hydrogen bond donors/acceptors.
- Electronic Descriptors: Energy of the highest occupied molecular orbital (HOMO), energy of the lowest unoccupied molecular orbital (LUMO).
- Nanomaterial Descriptors: Particle size, zeta potential, surface functional group density.
- Dataset Curation: Compile a training dataset where these features are linked to experimental outcomes, such as the limit of detection (LOD), sensitivity, or selectivity for a specific NSAID.
- Model Training and Validation:
- Algorithm Selection: Train a suite of ML algorithms, such as Random Forest, Support Vector Machines, or Gradient Boosting, on the curated dataset. For larger datasets, deep learning architectures like multilayer perceptrons (MLPs) can be employed [77].
- Validation: Use k-fold cross-validation to assess model performance and avoid overfitting. The model's goal is to learn the complex relationship between the input features and the sensor performance metrics.
- Virtual Screening and Prediction: Deploy the trained model to screen a vast virtual library of potential sensor materials or structures. The model predicts the performance of each virtual candidate, prioritizing the most promising ones for synthesis and experimental validation.
- Active Learning and Iteration: Incorporate the experimental results from the newly tested candidates back into the training dataset. This iterative process, known as active learning, continuously improves the model's predictive accuracy and guides the exploration of the chemical space more efficiently [81] [82].
Workflow Visualization: Integrated HTS and ML for Sensor Development
The following diagram illustrates the synergistic, iterative workflow that integrates high-throughput experimentation with machine learning, creating a closed-loop system for accelerated sensor development.
Integrated HTS and ML Workflow for Sensor Development
The Scientist's Toolkit: Essential Research Reagents and Materials
The table below details key reagents, materials, and instrumentation essential for implementing the HTS and ML-assisted workflows described in this guide.
Table 2: Essential Research Tools for HTS and ML-Assisted Sensor Development
| Tool Category | Specific Examples | Function in NSAID Sensor Research |
|---|---|---|
| Nanomaterial Modifiers [78] [76] | Graphene oxide, Carbon nanotubes (CNTs), Metal nanoparticles (Au, Ag), MXenes (Ti₃C₂Tₓ). | Enhance electrode conductivity, surface area, and catalytic activity, leading to improved sensitivity and lower detection limits for NSAIDs. |
| Recognition Elements [12] [76] | Molecularly Imprinted Polymers (MIPs), Aptamers, Anthracene/naphthalimide-based fluorescent receptors. | Provide selective binding sites for specific NSAIDs, reducing cross-reactivity and improving sensor specificity in complex samples. |
| HTS Hardware [79] [83] | Robotic liquid handlers, 1536-well microplates, Automated plate readers (electrochemical/optical). | Enable the miniaturization, automation, and parallel processing required to screen thousands of sensor conditions rapidly and reproducibly. |
| Electrochemical Techniques [78] [76] | Cyclic Voltammetry (CV), Differential Pulse Voltammetry (DPV), Electrochemical Impedance Spectroscopy (EIS). | Transduce the binding or redox event of an NSAID at the sensor surface into a quantifiable electrical signal. DPV is prized for its high sensitivity. |
| Data Processing Tools [79] [80] | R/Python with packages for robust statistical analysis (e.g., for trimmed-mean polish), Hill equation fitting algorithms. | Correct for systematic experimental noise and model concentration-response data to extract reliable performance parameters (AC~50~, E~max~). |
| ML Software & Libraries [77] [82] | Scikit-learn, PyTorch, TensorFlow, PyMC. | Provide the algorithms and infrastructure for building, training, and validating models that predict sensor performance and guide experimental design. |
The integration of high-throughput screening and machine learning is no longer a futuristic concept but a present-day reality that is actively reshaping the landscape of sensor characterization and development for NSAIDs. As these technologies mature, several key trends are poised to define their future impact. The push for automation, extending from assay execution to data analysis, will further unify and accelerate the research workflow, making it more reproducible and efficient [82]. The development of next-generation electrochemical and optical sensors will increasingly rely on hybrid nanomaterials and sophisticated data analysis to achieve sub-micromolar detection limits in real-world samples, moving analysis from central laboratories to the point-of-care [78] [76].
However, for this potential to be fully realized, several challenges must be addressed. The field requires larger, standardized, and high-quality datasets to train more robust and generalizable ML models. Furthermore, bridging the interdisciplinary gap between data scientists and experimental researchers is crucial for designing disciplined digital systems from the ground up [82]. By embracing these integrated approaches, the research community can accelerate the design of highly sensitive and selective sensors, not only advancing pharmaceutical analysis and therapeutic drug monitoring but also strengthening our capabilities in environmental surveillance and personalized medicine.
Conclusion
Effectively characterizing and mitigating non-specific adsorption is paramount for the development of reliable biosensors for clinical and environmental applications. A foundational understanding of NSA mechanisms informs the strategic selection of characterization techniques, such as the powerful combination of electrochemical and SPR methods. The ongoing development of advanced antifouling materials, coupled with robust validation frameworks and the emerging use of machine learning, paves the way for a new generation of biosensors. Future research must focus on creating standardized protocols and scalable, reproducible coatings to translate lab-scale successes into real-world diagnostic and monitoring tools, ultimately enhancing their impact on biomedical research and public health.
References
- 1. Promising Solutions to Address the Non-Specific ... [https://www.mdpi.com/2227-9040/13/3/92]
- 2. Non-Specific Adsorption Reduction Methods in Biosensing [https://www.semanticscholar.org/paper/Non-Specific-Adsorption-Reduction-Methods-in-Lichtenberg-Ling/498c19ce009e78ec3a6f75ba09e8b3c7be162139]
- 3. – Adsorption and Physisorption Explained Chemisorption [https://www.gcea.de/en/artikel/adsorption]
- 4. A quantitative method to discriminate between non - specific and... [https://pubmed.ncbi.nlm.nih.gov/26619130/]
- 5. meaning- Adsorption application | Adsorption types Adsorption [https://allen.in/jee/chemistry/adsorption]
- 6. AI-Enhanced Surface Functionalization in Biosensors [https://www.sciencedirect.com/science/article/abs/pii/S0165993625003887]
- 7. Porous silicon biosensors meet zwitterionic peptides [https://pubs.rsc.org/en/content/articlehtml/2025/nh/d5nh00478k]
- 8. MOF nanozymes: active sites and sensing applications [https://pubs.rsc.org/en/content/articlehtml/2025/qi/d4qi02555e]
- 9. Towards chemical accuracy for chemi- and physisorption with an... [https://arxiv.org/html/2410.11248v2]
- 10. Understanding the selectivity of nonsteroidal - anti ... inflammatory [https://pmc.ncbi.nlm.nih.gov/articles/PMC11878451/]
- 11. ES-Screen: A Novel Electrostatics-Driven Method for Drug ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9736079/]
- 12. Advances in detecting non-steroidal anti-inflammatory drugs ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11605430/]
- 13. Diclofenac and other non-steroidal anti-inflammatory drugs ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10060569/]
- 14. NMR Investigation of the Interaction of Three Non-Steroidal ... [https://www.mdpi.com/1420-3049/27/19/6647]
- 15. Quantitative structure activity relationships studies of non- ... [https://www.sciencedirect.com/science/article/pii/S1878535216300168]
- 16. Metal-Based Nanomaterials for the Sensing of NSAIDS [https://www.mdpi.com/2079-4991/14/7/630]
- 17. Non-steroidal anti-inflammatory drugs (NSAIDs) in the ... [https://www.sciencedirect.com/science/article/pii/S0147651323011284]
- 18. Non-Specific Adsorption Reduction Methods in Biosensing [https://pmc.ncbi.nlm.nih.gov/articles/PMC6603772/]
- 19. Machine learning assisted wearable antifouling sensor for ... [https://www.sciencedirect.com/science/article/abs/pii/S0026265X25025305]
- 20. Reduction of Biosensor False Responses and Time Delay ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10683760/]
- 21. “Jack-of-All-Trades” properties of nanozyme opens up an ... [https://www.sciencedirect.com/science/article/abs/pii/S1385894725089557]
- 22. PFAS Levels in Paired Drinking Water and Serum Samples ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10529814/]
- 23. Associations between PFAS in public water system ... [https://www.nature.com/articles/s41370-025-00817-8]
- 24. Startup's biosensor makes drug development and ... [https://news.mit.edu/2025/startup-biosensor-makes-drug-development-and-manufacturing-cheaper-0615]
- 25. what is Electrochemical Methods? [https://www.zensorrd.com/Article10.html]
- 26. Key Electrochemical Techniques Every Researcher Should ... [https://www.msesupplies.com/blogs/news/key-electrochemical-techniques-every-researcher-should-know?srsltid=AfmBOopkO9tFnpMpz4gEBOa52F8uzknun93_M5td-alQrxMl65PljeIO]
- 27. Synthesis and characterization of a highly sensitive and ... [https://www.sciencedirect.com/science/article/abs/pii/S0039914020302447]
- 28. Strategies to Enrich Electrochemical Sensing Data with ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11207790/]
- 29. Comparison of cyclic voltammetry measurements of paper ... [https://bioresources.cnr.ncsu.edu/resources/comparison-of-cyclic-voltammetry-measurements-of-paper-based-screen-printed-electrodes-via-proprietary-and-open-source-potentiostat/]
- 30. 表面等离激元增强的光和物质相互作用 [https://wulixb.iphy.ac.cn/cn/article/doi/10.7498/aps.68.20190337]
- 31. 表面等离子共振法(SPR)测定抗体亲和力-Biacore工作原理 [https://www.detaibio.com/topics/surface-plamon-resonace-technology.html]
- 32. 表面等离子共振仪( SPR ) [https://www.bruker.com/zh/products-and-solutions/surface-plasmon-resonance.html]
- 33. 近30亿潜在市场规模,新一代SPR技术检测平台市场研究报告 [https://www.sohu.com/a/382899424_133140]
- 34. 离焦对表面等离激元共振显微成像的影响 [https://www.opticsjournal.net/Articles/OJ524af519cee10b50/FullText]
- 35. Electrochemistry-coupled surface plasmon resonance on ... [https://www.sciencedirect.com/science/article/pii/S2451910324001959]
- 36. Simultaneous Voltammetric Determination of Non-Steroidal ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9823404/]
- 37. Simultaneous Quantification of Four Principal NSAIDs ... [https://www.mdpi.com/2673-4583/5/1/3]
- 38. Ultrasensitive and simultaneous detection of 6 nonsteroidal ... [https://www.sciencedirect.com/science/article/pii/S0022030221000588]
- 39. Surface Plasmon Resonance (SPR)-Based Workflow for High ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12612902/]
- 40. Self-assembly strategy to reduce non-specific adsorption ... [https://www.sciencedirect.com/science/article/abs/pii/S0003267022009382]
- 41. Advances and Prospects of Nanomaterial Coatings in ... [https://www.mdpi.com/2079-6412/15/9/1008]
- 42. Polymer Coatings Market Growth 2025, Opportunities and ... [https://www.einpresswire.com/article/866186370/polymer-coatings-market-growth-2025-opportunities-and-leading-players-analysis-forecast-to-2031]
- 43. Polymer Coatings Market Share, Growth Trends 2026-2035 [https://www.researchnester.com/reports/biopolymer-coatings-market/7159]
- 44. Polymer Emulsions Market | Global Market Analysis Report [https://www.futuremarketinsights.com/reports/polymer-emulsions-market]
- 45. Polymer-Nanoparticle Composites: From Synthesis to ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5521756/]
- 46. Analytical Methods for Characterization of Nanomaterial ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7941215/]
- 47. U.S. Specialty Polymers Market Size to Surpass USD 59.52 ... [https://www.towardschemandmaterials.com/insights/us-specialty-polymers-market]
- 48. A Review of Recent Progress in Synthetic Polymer Surface ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12250919/]
- 49. a review focusing on active and passive anti-fouling strategies [https://pubs.rsc.org/en/content/articlehtml/2025/ta/d5ta03722k]
- 50. Bioinspired coating with active-passive synergistic ... [https://www.sciencedirect.com/science/article/abs/pii/S0300944025002292]
- 51. Supramolecular dynamics-enhanced synergistic ... [https://www.nature.com/articles/s41467-025-62231-w]
- 52. Evaluation of the effectiveness and durability of commercial ... [https://www.sciencedirect.com/science/article/pii/S0260877424000256]
- 53. Experimental investigation on the hydrodynamic ... [https://www.sciencedirect.com/science/article/abs/pii/S0029801821007253]
- 54. Controlling surface porosity of graphene-based printed ... [https://www.nature.com/articles/s41699-022-00312-w]
- 55. Electrospun Nanofiber Membrane: Charge Characteristics ... [https://www.sciencedirect.com/science/article/pii/S1383586625036822]
- 56. Progress in modified electrospun PVC membranes [https://www.sciencedirect.com/science/article/abs/pii/S1383586624041959]
- 57. Long-term drift behavior in metal oxide gas sensor arrays [https://www.nature.com/articles/s41597-025-05993-8]
- 58. Exploring Wearable Sensors for Therapeutic Drug Monitoring ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10377150/]
- 59. Comparing the Long-Term Stability and Measurement ... [https://www.mdpi.com/2227-9717/13/10/3306]
- 60. Do AirGradient PM2.5 Monitors Stay Accurate Over Time? ... [https://www.airgradient.com/blog/do-our-pm2.5-sensors-drift/]
- 61. An adversarial network used for drift correction in electronic ... [https://www.sciencedirect.com/science/article/abs/pii/S0924424724007143]
- 62. Inhibition of non-specific protein adsorption on PMMA surface [https://www.sciencedirect.com/science/article/pii/S131961032300159X]
- 63. Nano structured sensing surface: Significance in sensor ... [https://www.sciencedirect.com/science/article/abs/pii/S0925400518307895]
- 64. Understanding Sensitivity in Nanoscale Sensing Devices [https://pmc.ncbi.nlm.nih.gov/articles/PMC12183584/]
- 65. Emerging optical nanoscopy techniques | NSA [https://www.dovepress.com/emerging-optical-nanoscopy-techniques-peer-reviewed-fulltext-article-NSA]
- 66. An Improved Method for Quantitatively Measuring ... [https://www.pnnl.gov/publications/improved-method-quantitatively-measuring-antifouling-coating-performance-using-mussel]
- 67. An improved method for quantitatively measuring ... [https://pubmed.ncbi.nlm.nih.gov/40103291/]
- 68. A Method for Evaluating the Efficacy of Antifouling Paints ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5154544/]
- 69. ISO standard for performance evaluation of antifouling ... [https://smartmaritimenetwork.com/2025/10/01/iso-standard-for-performance-evaluation-of-antifouling-paints-published/]
- 70. Assessment of the effectiveness of antifouling solutions for ... [https://www.sciencedirect.com/science/article/pii/S0025326X24000857]
- 71. Evaluation of the Anti-fouling Efficacy of Bacillus ... [https://www.frontiersin.org/journals/marine-science/articles/10.3389/fmars.2021.711108/full]
- 72. Electrochemistry combined-surface plasmon resonance ... [https://www.sciencedirect.com/science/article/pii/S0165993622002497]
- 73. Self-referencing SPR biosensing with an ultralow limit-of- ... [https://www.sciencedirect.com/science/article/pii/S092540052031282X]
- 74. Quantitative Evaluation of Sensitivity and Selectivity ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC2072052/]
- 75. Analytical methods for quantification of non-steroidal anti ... [https://www.sciencedirect.com/science/article/pii/S1878535223009085]
- 76. Recent Advances in Electrochemical Sensors for the ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12563010/]
- 77. Transforming modern drug discovery with machine learning [https://www.sciencedirect.com/science/article/abs/pii/S0065774325000016]
- 78. Recent developments in electrochemical sensors for the ... [https://www.sciencedirect.com/science/article/pii/S0013468625017499]
- 79. Quantitative high-throughput screening data analysis [https://pmc.ncbi.nlm.nih.gov/articles/PMC4375054/]
- 80. Experimental Design and Statistical Methods for Improved ... [https://www.sciencedirect.com/science/article/pii/S2472555222079801]
- 81. High-throughput screening accelerated by machine ... [https://pubs.rsc.org/en/content/articlelanding/2025/nr/d5nr00587f]
- 82. Making science run at the speed of thought: the reality of AI ... [https://www.drugtargetreview.com/article/190638/making-science-run-at-the-speed-of-thought-the-reality-of-ai-in-drug-discovery-part-2/]
- 83. High Throughput Screening Market Market 2025: Artificial ... [https://www.linkedin.com/pulse/high-throughput-screening-market-2025-artificial-4x9vf/]