CRISPR Biosensors: Revolutionizing Microbial Detection for Safer, Smarter Food Fermentation
This article explores the transformative potential of CRISPR-based biosensors for monitoring microbial communities in food fermentation processes.
CRISPR Biosensors: Revolutionizing Microbial Detection for Safer, Smarter Food Fermentation
Abstract
This article explores the transformative potential of CRISPR-based biosensors for monitoring microbial communities in food fermentation processes. Tailored for researchers and scientists, it provides a comprehensive analysis spanning the foundational principles of CRISPR-Cas systems, their practical application in detecting pathogens and starter cultures, optimization strategies for complex food matrices, and a critical comparison with traditional diagnostic methods. By synthesizing recent advancements and current challenges, this review aims to serve as a strategic guide for integrating these highly specific, sensitive, and portable diagnostic tools into food safety and quality control frameworks, ultimately paving the way for more resilient and data-driven fermentation industries.
The CRISPR-Cas Foundation: From Bacterial Immunity to Fermentation Monitoring
Core Mechanism of Adaptive Immunity
The Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR) and CRISPR-associated (Cas) proteins constitute an adaptive immune system that confers resistance to foreign genetic elements in prokaryotes, including plasmids and phages [1] [2]. This system provides a DNA-encoded, RNA-mediated, and sequence-specific adaptive defense mechanism [3]. Conceptually, CRISPR-Cas immunity shares functional parallels with the mammalian adaptive immune system while exhibiting characteristics of Lamarckian evolution, as acquired immunological memories are integrated into the host genome and vertically inherited [1].
Immunization occurs through a three-stage process, summarized in Table 1, which allows prokaryotic cells to adapt to new infectious threats and mount a targeted response upon subsequent encounters [1] [4].
Table 1: The Three Stages of CRISPR-Cas Adaptive Immunity
| Stage | Key Function | Primary Components | Molecular Outcome |
|---|---|---|---|
| Adaptation | Acquisition of new immunological memories | Cas1, Cas2, Protospacer Adjacent Motif (PAM) | Integration of a short viral or plasmid DNA sequence (protospacer) as a new spacer into the CRISPR locus [1] [4]. |
| crRNA Biogenesis | Expression of CRISPR guides | CRISPR locus, Cas proteins, RNases | Transcription of the CRISPR locus into a long precursor RNA, processed into short, mature CRISPR RNAs (crRNAs) [4]. |
| Interference | Target degradation and neutralization | crRNA, Cas effector nuclease (e.g., Cas9, Cas12) | crRNAs guide Cas nucleases to complementary invading nucleic acids, leading to their specific cleavage and degradation [1] [2]. |
Experimental Workflow for Studying CRISPR-Cas Function
The following diagram and protocol outline a generalized experimental approach for investigating the CRISPR-Cas immune response, from the initial immunization event to the assessment of immunity.
Protocol 1: Investigating Spacer Acquisition and Immunity
Objective: To demonstrate the adaptive acquisition of spacers from an invasive phage and confirm the resulting immunity.
Materials:
- Bacterial Strain: e.g., Streptococcus thermophilus DGCC7710 (a model strain with well-characterized CRISPR loci) [1].
- Bacteriophage: A virulent phage specific to the host strain.
- Growth Media: Appropriate broth and agar media (e.g., M17 broth with lactose).
- PCR Reagents: DNA polymerase, dNTPs, primers flanking the CRISPR locus.
- Gel Electrophoresis System: For analyzing PCR products.
- Sequencing Reagents: For verifying spacer sequences.
Procedure:
- Immunization Challenge: Grow the bacterial culture to mid-log phase. Infect with the bacteriophage at a high multiplicity of infection (MOI > 3) to ensure a high probability of infection.
- Selection and Isolation: After lysis is observed, plate the culture on solid media to isolate surviving colonies. Incubate under optimal conditions.
- Genotypic Analysis:
- Pick resistant colonies and inoculate into liquid culture.
- Extract genomic DNA from these cultures and a naive control culture.
- Use PCR with primers specific to the CRISPR locus to amplify the region.
- Analyze the PCR products by gel electrophoresis. A successful spacer acquisition will result in a larger PCR product compared to the control.
- Sequence the amplified CRISPR locus to confirm the identity of the newly acquired spacer, which should be homologous to a sequence (protospacer) in the bacteriophage genome [1].
- Phenotypic Confirmation: Challenge the immunized strain (and a naive control) with the same bacteriophage. Use an efficiency of plaquing (EOP) assay to quantify the survival rate. A significant reduction in EOP indicates successful, sequence-specific immunity [1].
Application in Biosensing: Detection of Foodborne Pathogens
The programmable nucleic acid recognition capability of CRISPR-Cas systems has been repurposed for highly sensitive and specific diagnostic tools, such as detecting pathogens in food fermentation research and safety monitoring [5] [6]. Systems utilizing Cas12a and Cas13a are particularly valuable due to their "collateral cleavage" activity, which allows for signal amplification.
Key CRISPR Effector Proteins for Biosensing
Table 2: Key Cas Effector Proteins Used in Biosensing Applications
| Effector Protein | Type | Target | PAM / PFS | Trans-Cleavage Activity | Key Feature for Detection |
|---|---|---|---|---|---|
| Cas9 | II | dsDNA | 5'-NGG-3' | No | Specific cleavage of target DNA; used with dCas9 for binding-based detection without cleavage [6] [7]. |
| Cas12a (Cpf1) | V | ds/ssDNA | 5'-TTTN-3' | Yes, ssDNA | Upon target recognition, cleaves non-specific single-stranded DNA (ssDNA) reporters, enabling highly sensitive signal amplification [6] [7]. |
| Cas13a | VI | ssRNA | 3' non-G PFS | Yes, ssRNA | Upon target recognition, cleaves non-specific single-stranded RNA (ssRNA) reporters. Ideal for detecting RNA viruses or transcriptional activity [6]. |
The mechanism of a typical Cas12a-based biosensor is illustrated below.
Protocol 2: CRISPR-Cas12a Biosensor for DetectingSalmonella typhimurium
Objective: To sensitively and specifically detect S. typhimurium in a sample using a Cas12a-based assay with a colorimetric or fluorescent readout [8].
Materials:
- Cas12a Protein: Recombinantly expressed and purified.
- crRNA: Designed to be specific to a unique sequence in the S. typhimurium genome.
- ssDNA Reporter: For fluorescent detection: an ssDNA oligonucleotide with a fluorophore (e.g., FAM) and a quencher (e.g., BHQ1). For colorimetric detection: a system like a multi-indicator pH millidisc that responds to pH changes induced by the reaction byproducts [8].
- Isothermal Amplification Kit: e.g., Recombinase Polymerase Amplification (RPA) or Recombinase-Aided Amplification (RAA) for amplifying target DNA without a thermal cycler.
- Buffer: 10× NEBuffer for the Cas12a reaction.
- Signal Detection Device: Fluorescence plate reader, or a smartphone-based imaging platform for colorimetric analysis [8].
Procedure:
- Sample Preparation and Amplification:
- Extract DNA from the food sample (e.g., chicken homogenate).
- Amplify the target DNA using an isothermal amplification method (RPA/RAA) according to the kit protocol.
- CRISPR-Cas12a Reaction Setup:
- Prepare the detection mix:
- 10 μL of enzyme-free sterile water
- 10 μL of 10 × NEBuffer
- 10 μL of 1 μM Cas12a
- 10 μL of 1 μM crRNA
- 5 μL of 1 μM ssDNA fluorescent reporter
- Incubate at 25°C for 10 minutes to allow the Cas12a-crRNA complex to form.
- Add 5 μL of the amplified DNA product (or non-target control) to the mixture.
- Incubate the entire reaction at 37°C for 30-60 minutes.
- Prepare the detection mix:
- Signal Readout:
- Fluorescent Detection: Measure the fluorescence intensity using a plate reader. A positive sample will show a significant increase in fluorescence compared to the control.
- Colorimetric Detection (Alternative): If using a pH-sensitive system, the cleavage reaction can be coupled to an enzyme that produces a pH change. The color shift can be visualized and quantified using a smartphone app [8].
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Reagent Solutions for CRISPR-Cas Research and Biosensing
| Research Reagent | Function | Example Use Case | Critical Notes |
|---|---|---|---|
| Cas Effector Proteins (e.g., Cas9, Cas12a, Cas13a) | The core enzymatic component that executes nucleic acid cleavage. | Cas12a is used in DETECTR for DNA virus detection; Cas13a is used in SHERLOCK for RNA virus detection [5] [6]. | Specificity and activity vary by type. Cas12a requires a T-rich PAM, while Cas9 requires an NGG PAM. |
| Guide RNAs (crRNA, sgRNA) | Provides sequence specificity by guiding the Cas protein to the target nucleic acid. | A crRNA is designed with a 20-nt spacer complementary to a unique gene in Listeria monocytogenes for specific detection [9] [6]. | The seed sequence (8-10 bases at the 3' end of the guide) is critical for specificity and intolerant to mismatches. |
| Protospacer Adjacent Motif (PAM) | A short, specific DNA sequence adjacent to the target protospacer; essential for Cas protein recognition. | When designing a Cas9 assay, the target sequence must be directly adjacent to a 5'-NGG-3' PAM [1] [9]. | PAM sequence requirements are a key differentiator between Cas proteins and a primary constraint in assay design. |
| Reporter Probes (ssDNA, ssRNA) | A labeled nucleic acid strand that is non-specifically cleaved upon Cas activation, generating a detectable signal. | A FAM-quenched ssDNA reporter is cleaved by activated Cas12a, producing a fluorescent signal for real-time detection [8] [6]. | The reporter is the basis for signal amplification in Cas12 and Cas13 systems, enabling high sensitivity. |
| Isothermal Amplification Kits (RPA, RAA) | Amplifies target nucleic acids to detectable levels at a constant temperature, enabling portable detection. | Used to pre-amplify target DNA from Salmonella in a food sample before Cas12a detection, boosting sensitivity to attomolar levels [5]. | Eliminates the need for expensive thermal cyclers, making the biosensor suitable for field deployment. |
Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR) and CRISPR-associated (Cas) systems provide adaptive immunity in bacteria and archaea, and their effector proteins have been repurposed as revolutionary tools in biotechnology and molecular biology [10] [11]. These systems are broadly classified into two classes: Class 1 (types I, III, and IV) utilizes multi-subunit effector complexes, while Class 2 (types II, V, and VI) employs single, multi-domain effector proteins, making them particularly amenable for technological applications [10] [12]. Among Class 2, the key effector proteins are Cas9 (type II), Cas12 (type V), and Cas13 (type VI) [13]. This application note provides a detailed classification and structural breakdown of these effectors, framed within their application in developing CRISPR-based biosensors for microbial detection in food fermentation research. Understanding their distinct domains, mechanisms, and requirements is crucial for researchers aiming to deploy these precision tools for ensuring food safety and quality.
Classification and Comparative Analysis of Cas Effectors
The core Class 2 CRISPR-Cas effectors—Cas9, Cas12, and Cas13—share a common bilobed architecture but diverge significantly in their target specificity, nuclease domains, and activation mechanisms. Table 1 provides a comprehensive comparison of their defining characteristics.
Table 1: Structural and Functional Characteristics of Class 2 CRISPR-Cas Effectors
| Feature | Cas9 | Cas12a | Cas13a |
|---|---|---|---|
| Class 2 Type | Type II [10] | Type V [10] | Type VI [13] |
| Target Nucleic Acid | Double-stranded DNA (dsDNA) [10] | dsDNA / Single-stranded DNA (ssDNA) [10] | Single-stranded RNA (ssRNA) [10] |
| gRNA Composition | crRNA + tracrRNA, or single-guide RNA (sgRNA) [10] [14] | crRNA [10] or crRNA + tracrRNA (for Cas12c) [15] | crRNA [13] |
| gRNA Size | ~100 nt [10] | ~40 nt [10] | ~50 nt [10] |
| Spacer Position | 5′ spacer [10] | 3′ spacer [10] | 3′ spacer [10] |
| PAM / PFS Requirement | Yes (e.g., NGG for SpCas9) [10] [14] | Yes (e.g., TTTV for Cas12a) [10] [14] | No PAM; requires Protospacer Flanking Site (PFS) [13] |
| Nuclease Domain(s) | HNH & RuvC [10] [14] | Single RuvC domain [10] [15] | Two HEPN domains [10] [13] |
| Cleavage Activity | Cleaves both DNA strands (cis) [10] | Cleaves both DNA strands (cis); exhibits collateral trans-cleavage of ssDNA [10] | Cleaves target RNA (cis); exhibits collateral trans-cleavage of ssRNA [10] [13] |
| pre-crRNA Processing | Requires host RNase III and tracrRNA [14] | Cas12 effectors are typically self-processing [15] | Cas13 effectors are self-processing [13] |
The following diagram illustrates the fundamental mechanisms and nucleic acid targeting of these three key effectors.
Domain Architecture and Molecular Mechanisms
Cas9: The DNA Double-Strand Breaker
Cas9 proteins exhibit a conserved bilobed architecture composed of a Recognition (REC) lobe and a Nuclease (NUC) lobe [10]. The REC lobe (comprising REC1, REC2, and REC3 domains) is primarily responsible for binding the guide RNA and facilitating the recognition of the target DNA sequence [10]. The NUC lobe contains the two nuclease domains and the PAM-interacting (PI) domain.
- HNH Domain: Cleaves the DNA strand complementary to the guide RNA (target strand) [10] [14].
- RuvC Domain: Cleaves the non-complementary DNA strand (non-target strand) [10] [14].
- PI Domain: Critical for recognizing the short Protospacer Adjacent Motif (PAM) sequence in the target DNA (e.g., 'NGG' for S. pyogenes Cas9), which is a primary conformational checkpoint for activation [10].
Activation of Cas9 is a multi-step process gated by conformational checkpoints. Upon PAM recognition, the DNA is partially unwound, allowing for seed sequence interrogation. Subsequent full R-loop formation (stable hybridization between the guide RNA and the target DNA) triggers a large conformational shift in the REC lobe, which allosterically drives the HNH domain to pivot into an active configuration for cleavage, which in turn activates the RuvC domain [10].
Cas12: The DNA-Targeting, Single-RNase Effector
The Cas12 family is highly diverse, including Cas12a (Cpf1), Cas12c, and miniature variants like Cas12f, but all share a single RuvC nuclease domain and lack an HNH domain [15] [16]. Their general architecture also includes REC and NUC lobes.
- RuvC Domain: The sole nuclease domain responsible for cleaving both strands of the target dsDNA [10] [15]. It is also used by some Cas12 variants (e.g., Cas12c2) for processing their own precursor crRNA [15].
- REC Lobe: Facilitates guide RNA and target DNA binding.
- WED Domain: Within the NUC lobe, it plays a key role in nucleic acid binding.
- PI Domain: Recognizes a T-rich PAM (e.g., 'TTTV' for Cas12a, 'TN' for Cas12c2) [10] [15].
A defining feature of many Cas12 effectors (like Cas12a) is their collateral cleavage activity. Upon formation of a ternary complex with a target dsDNA, the Cas12 RuvC domain becomes a nonspecific deoxyribonuclease that cleaves nearby single-stranded DNA (ssDNA) molecules [10] [5]. This trans-cleavage activity, which is highly activated in biosensing applications, continues as long as the effector is target-bound.
Cas13: The RNA-Targeting, Collateral RNase
Cas13 effectors specialize in targeting and cleaving single-stranded RNA and are characterized by the presence of two Higher Eukaryotes and Prokaryotes Nucleotide-binding (HEPN) domains [13]. Similar to other Class 2 effectors, they possess a bilobed (REC-NUC) structure.
- HEPN Domains: Two conserved HEPN domains (HEPN1 and HEPN2) form the active site for RNA cleavage. In the inactive state, these domains are separated. Target RNA binding induces a conformational change that brings the two HEPN domains into proximity, creating a functional RNase active site [13] [17].
- REC Lobe: Composed of N-terminal and Helical-1 domains, it is responsible for crRNA recognition and binding [13].
- NUC Lobe: Contains the Helical-2 domain and the two HEPN domains.
Like Cas12, Cas13 exhibits robust collateral cleavage activity. Target binding activates the HEPN domains, enabling them to promiscuously cleave any surrounding non-target ssRNA molecules [13] [5]. This property is harnessed in sensitive RNA detection platforms. Furthermore, Cas13 proteins are self-processing and mature their own precursor crRNAs without the need for host factors [13].
Experimental Protocols for Biosensor Development
The following protocol outlines the development of a CRISPR-based biosensor for detecting specific microbial contaminants or monitoring starter cultures in food fermentation, leveraging the collateral activity of Cas12 or Cas13.
Protocol: CRISPR-Cas12/Cas13-based Fluorescent Detection of Microbial Nucleic Acids
Principle: Target DNA/RNA from a microbial source is amplified isothermally. The amplicon then activates the collateral cleavage activity of Cas12/Cas13, which cleaves a fluorescently quenched reporter probe, generating a fluorescent signal.
I. Sample Preparation and Nucleic Acid Amplification
- Sample Lysis: Homogenize the food sample (e.g., 1 g of fermented meat or dairy product) in a lysis buffer. Use mechanical disruption (e.g., bead beating) for robust cell lysis.
- Nucleic Acid Extraction: Extract total nucleic acid using a commercial kit. For RNA-specific targets (using Cas13), include a DNase I treatment step.
- Isothermal Amplification:
- Perform Recombinase Polymerase Amplification (RPA) for DNA targets or Reverse Transcription-RPA (RT-RPA) for RNA targets.
- Reaction Setup:
- 29.5 µL of rehydration buffer (with primers).
- 2 µL of template nucleic acid.
- 1 µL of fluorescent reporter probe (e.g., 10 µM ssDNA-FQ for Cas12; ssRNA-FQ for Cas13).
- Add magnesium acetate to a final concentration of 14 mM to start the reaction.
- Incubate at 37-42°C for 15-30 minutes.
II. CRISPR-Cas Detection Reaction
- Reaction Mix Preparation (per reaction):
- 5 µL of Nuclease-Free Water.
- 2 µL of 10X Cas Reaction Buffer (e.g., 200 mM HEPES, 1M NaCl, pH 6.5).
- 2 µL of purified Cas12 or Cas13 protein (e.g., 100-500 nM final concentration).
- 2 µL of guide RNA (crRNA for Cas12, crRNA for Cas13) (e.g., 100-500 nM final concentration).
- Assay Assembly:
- Transfer 2 µL of the isothermal amplification product directly into the prepared CRISPR reaction mix.
- Mix gently by pipetting.
- Incubate at 37°C for 10-30 minutes.
III. Signal Measurement and Analysis
- Real-time Fluorescence Monitoring: Use a plate reader or portable fluorimeter to measure fluorescence (FAM channel: Ex 485 nm, Em 520 nm) at 1-minute intervals during incubation.
- Endpoint Visualization: After incubation, visualize results under a blue light transilluminator. A positive sample will fluoresce brightly, while a negative sample will remain dark.
- Quantification (Optional): Generate a standard curve using known concentrations of the target nucleic acid to estimate the load of the microbe in the original sample.
The workflow for this protocol is summarized in the following diagram.
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Reagents for CRISPR-based Microbial Biosensing
| Reagent / Material | Function / Description | Example Application / Note |
|---|---|---|
| Cas Effector Protein | The core enzyme for programmable nucleic acid recognition and cleavage. | Purified recombinant Cas12a (for DNA) or Cas13a (for RNA). Lyophilized, cold-chain stable proteins are ideal for field use [5]. |
| Guide RNA (crRNA) | Provides target specificity by guiding the Cas effector to the complementary sequence. | Chemically synthesized crRNA designed against a unique microbial genomic region (e.g., a virulence gene or species-specific marker) [5]. |
| Isothermal Amplification Kit | Amplifies target nucleic acid to detectable levels at a constant temperature. | RPA kit (for DNA) or RT-RPA kit (for RNA). Enables rapid amplification without the need for a thermal cycler [5]. |
| Fluorescent Reporter Probe | A quenched nucleic acid substrate cleaved during collateral activity, producing fluorescence. | For Cas12: ssDNA oligonucleotide with 5'-fluorophore/3'-quencher (e.g., FAM/TAMRA). For Cas13: ssRNA equivalent [5]. |
| Portable Fluorimeter | Instrument for measuring real-time or endpoint fluorescence. | Essential for quantitative results. Compact, battery-operated devices enable on-site testing in production facilities [18] [5]. |
| Nucleic Acid Extraction Kit | Isolates DNA and/or RNA from complex food matrices. | Must be optimized for the specific food type (e.g., dairy, meat, produce) to remove PCR inhibitors [18]. |
Application in Food Fermentation Research
The unique properties of Cas effectors make them ideal for addressing critical challenges in food fermentation microbiology.
- Monitoring Starter Cultures: Cas13-based biosensors can be designed to target specific rRNA or mRNA from beneficial bacteria like Lactobacillus or Streptococcus thermophilus, allowing for real-time monitoring of their metabolic activity and viability during fermentation, rather than just their presence [18].
- Rapid Pathogen Detection: Cas12-based assays can detect DNA from common foodborne pathogens such as Listeria monocytogenes, Salmonella spp., and E. coli O157:H7 in hours instead of days, directly in food matrices like cheese, yogurt, or fermented sausages [18] [5]. This enables rapid intervention and prevents outbreaks.
- Spoilage Microbe Identification: Biosensors targeting spoilage organisms like Pseudomonas spp. or yeasts can provide early warning of spoilage, reducing economic losses [18]. The ability to perform this monitoring on-site (e.g., in a production facility) is a significant advantage over traditional lab-based methods.
The precise classification of Cas effectors based on their domain architecture and molecular mechanisms is the foundation for their rational application. Cas9, with its dual DNA cleavage, remains a powerful gene-editing tool. In contrast, Cas12 and Cas13, with their programmable specificity and potent collateral cleavage activities, are pioneering a new paradigm in molecular diagnostics. For researchers in food fermentation, integrating these CRISPR effectors into robust biosensing platforms offers an unprecedented opportunity to enhance food safety, optimize fermentation processes, and ensure product quality through rapid, sensitive, and specific detection of microbial targets.
CRISPR-Cas systems have emerged as powerful tools for molecular diagnostics, with their programmable nucleic acid recognition capabilities offering unprecedented specificity and sensitivity. For researchers in food fermentation and microbial detection, understanding the distinct nuclease activities of CRISPR-Cas proteins—particularly cis-cleavage and trans-cleavage—is fundamental to developing effective biosensing strategies. These two mechanisms enable Cas proteins to function not only as precise molecular scissors but also as signal amplifiers, making them invaluable for monitoring microbial populations, detecting contaminants, and ensuring the safety and quality of fermented products [5] [19]. This application note details the mechanisms, comparative characteristics, and practical implementation of these activities within the context of food fermentation research.
Fundamental Mechanisms of Cas Protein Nuclease Activities
Cis-Cleavage: Targeted Sequence-Specific Cleavage
Cis-cleavage refers to the precise, targeted cutting of a specific nucleic acid sequence that is complementary to the CRISPR RNA (crRNA) guide. This activity is the foundation of the CRISPR system's adaptive immune function in bacteria and its initial application in gene editing.
- Molecular Mechanism: The Cas protein (e.g., Cas12a) forms a ribonucleoprotein complex with a crRNA. This complex scans DNA for a protospacer adjacent motif (PAM). Upon recognizing the PAM sequence, the crRNA hybridizes with the complementary target strand, triggering a conformational change in the Cas protein that activates its nuclease domains. For Cas12a, the RuvC domain is responsible for cleaving both strands of the target double-stranded DNA (dsDNA), generating a staggered double-strand break [20] [19].
- Key Characteristic: This cleavage is highly specific and is confined only to the target molecule that activated the enzyme.
Trans-Cleavage: Non-Specific Collateral Cleavage
Trans-cleavage, also known as collateral cleavage, is a non-specific nuclease activity that is unleashed after the Cas complex has bound and cleaved its target DNA or RNA via cis-cleavage.
- Molecular Mechanism: The activation of the Cas protein through successful cis-cleavage of its target induces a sustained enzymatic state. In this activated state, the protein indiscriminately cleaves any surrounding single-stranded DNA (for Cas12a) or single-stranded RNA (for Cas13a) molecules [20] [19]. This activity is often compared to a "shredding" mode.
- Key Characteristic: This activity is non-specific and operates in trans, meaning it degrades non-target molecules in the vicinity. This catalytic amplification is the cornerstone of CRISPR-based diagnostic biosensors.
The following diagram illustrates the sequential workflow of these two cleavage activities for a typical Cas12a system:
Comparative Analysis: Cis vs. Trans-Cleavage
The table below provides a structured comparison of the key features of cis- and trans-cleavage activities, essential for designing appropriate detection experiments.
Table 1: Comparative Analysis of Cis-Cleavage and Trans-Cleavage Activities in CRISPR-Cas Systems
| Feature | Cis-Cleavage | Trans-Cleavage (Collateral) |
|---|---|---|
| Definition | Sequence-specific cleavage of the target nucleic acid [19] | Non-specific, indiscriminate cleavage of surrounding non-target nucleic acids [19] |
| Specificity | High (requires complementarity to crRNA) | Low (cleaves non-target ssDNA or ssRNA indiscriminately) [20] |
| Function | Target recognition and initial activation | Signal amplification |
| Primary Role | Target identification | Signal generation and amplification for detection |
| Key Cas Proteins | Cas9, Cas12a, Cas13a | Cas12a (ssDNA), Cas13a (ssRNA) [20] |
| Output | Double-strand break in target DNA | Generation of measurable signal (e.g., fluorescence) |
| Kinetic Turnover | Single-turnover (one target per activation) | Multiple-turnover (catalytic, ~17 s⁻¹ for LbCas12a) [20] |
Quantitative Kinetics and Performance Data
The performance of CRISPR-based biosensors, particularly their sensitivity, is heavily dependent on the kinetics of the trans-cleavage reaction. Accurate measurement and optimization of these kinetics are critical for assay design.
Table 2: Key Kinetic and Performance Parameters for CRISPR-Cas Trans-Cleavage Activity
| Parameter | Value / Description | Significance / Notes |
|---|---|---|
| Reported Turnover Number (Kcat) | ~17 s⁻¹ for LbCas12a [20] | Earlier reports overestimated this value; corrected kinetics are essential for predicting LoD [20]. |
| Effect of crRNA Engineering | Up to 3.5-fold enhancement with 3' ssDNA extensions [21] | Engineered crRNAs (e.g., 7-mer DNA extension) can significantly boost trans-cleavage rates and sensitivity [21]. |
| Limit of Detection (LoD) with Pre-amplification | Attomolar (aM) to Femtomolar (fM) levels [22] | Combined with RPA or LAMP, enables single molecule detection. |
| Limit of Detection (LoD) Amplification-Free | Femtomolar (fM) to Picomolar (pM) levels [21] [23] | Engineered systems can achieve fM LoD without pre-amplification [21]; colorimetric systems report ~10 pM [23]. |
| Key Influencing Factors | Cas homolog, crRNA sequence, reporter composition, divalent cations, buffer conditions [20] | Optimization of these factors is required for maximal activity. |
Experimental Protocol: Detecting Microbial Targets via Cas12a Trans-Cleavage
This protocol details a method for detecting a specific microbial target (e.g., E. coli O157:H7) in a food sample using a CRISPR-Cas12a system coupled with isothermal pre-amplification and fluorescence readout [24] [19].
Research Reagent Solutions
Table 3: Essential Reagents and Materials for CRISPR-Cas12a Detection Assay
| Reagent/Material | Function / Description | Example / Source |
|---|---|---|
| LbCas12a or LbaCas12a Enzyme | The effector nuclease that performs cis- and trans-cleavage. | Commercially available (e.g., New England Biolabs) [24]. |
| Target-Specific crRNA | Guides Cas12a to the complementary DNA target sequence. | Synthesized commercially; can be engineered with 3' DNA extensions for enhanced activity [21]. |
| Fluorescent ssDNA Reporter | A short ssDNA oligo labeled with a fluorophore and quencher. Trans-cleavage separates the pair, generating a signal. | e.g., FAM-TTATT-3IABkFQ [21]. |
| Recombinase Polymerase Amplification (RPA) Kit | Isothermal nucleic acid amplification to increase the target concentration before CRISPR detection. | TwistAmp Basic Kit [24]. |
| Nucleic Acid Extraction Method | Isolates DNA from complex food matrices (e.g., fermented products). | FTA cards for simplified, equipment-free extraction [24] or commercial kits. |
| Buffer (NEBuffer) | Provides optimal ionic strength and pH for Cas12a activity. | Often supplied with the commercial Cas12a enzyme [24]. |
Detailed Step-by-Step Procedure
Step 1: Nucleic Acid Extraction from Food Sample
- Apply 100 µL of the food homogenate (e.g., fermented milk or meat slurry) onto an FTA card.
- Punch a 2 mm disc from the FTA card and transfer it to a microcentrifuge tube.
- Wash the disc sequentially with FTA purification reagent and TE buffer. Dry at 56°C for 10 min or at room temperature [24].
Step 2: Isothermal Pre-amplification (RPA)
- Prepare a 50 µL RPA reaction mixture containing:
- Rehydration buffer (29.5 µL)
- Forward and reverse primers (2 µL each, 240 nM final concentration)
- Reaction pellet
- Magnesium acetate (2.5 µL)
- Add the dried FTA disc containing the extracted DNA to the RPA mixture.
- Incubate the reaction at 37–42°C for 15–20 minutes to amplify the target DNA [24].
Step 3: CRISPR-Cas12a Detection Reaction
- Prepare the CRISPR detection mix in a separate tube:
- LbCas12a (100 nM final concentration)
- Target-specific crRNA (100 nM final concentration)
- Fluorescent ssDNA reporter (1 µM final concentration)
- 1x NEBuffer
- Add 1–2 µL of the RPA amplicon product to the CRISPR detection mix.
- Incubate the combined reaction at 37°C for 5–15 minutes.
Step 4: Signal Readout and Analysis
- Real-time Monitoring: Place the reaction tube in a real-time PCR instrument and measure the fluorescence every minute.
- Endpoint Analysis: Visualize the reaction tube under a blue-light transilluminator. A positive sample will show bright green fluorescence, while a negative control will remain dark.
- Quantification: Use standard curves to correlate the time-to-positive or endpoint fluorescence intensity with the original target concentration.
The complete workflow, from sample preparation to result interpretation, is summarized below:
Applications in Food Fermentation Research and Microbial Detection
The unique properties of cis- and trans-cleavage make CRISPR biosensors exceptionally suitable for addressing key challenges in food fermentation microbiology:
- Pathogen and Spoilage Organism Detection: Rapid detection of contaminants like Listeria monocytogenes, Salmonella spp., and E. coli O157:H7 directly in complex food matrices such as dairy, meat, and fresh produce with high specificity and sensitivity [18] [5]. For instance, an electrochemical biosensor utilizing Cas12a achieved a limit of detection of 19 CFU·mL⁻¹ for E. coli O157:H7 [25].
- Starter Culture Monitoring: Tracking the viability and activity of beneficial starter cultures (e.g., Lactobacillus spp., Streptococcus thermophilus) during fermentation to ensure process consistency and product quality [18].
- Real-time Process Control: Integration of CRISPR biosensors into smart fermentation platforms and IoT systems can provide real-time data on microbial populations, enabling dynamic process control and intervention to prevent batch failures [18] [26].
The CRISPR-Cas system, a cornerstone of modern genetic engineering, functions as a highly precise molecular toolkit for targeting and manipulating specific DNA sequences. Its operation hinges on the sophisticated interplay between its core components. In the context of microbial detection for food fermentation research, understanding these components is paramount for developing accurate and reliable biosensors. These biosensors can rapidly identify pathogens or monitor starter cultures, directly impacting food safety and quality [18]. The system fundamentally comprises a Cas nuclease and a guide RNA that directs it to a specific genomic locus. The target recognition process is further governed by a short DNA sequence known as the Protospacer Adjacent Motif (PAM) [9] [27]. This article will delve into the distinct roles of the CRISPR RNA (crRNA), the single guide RNA (sgRNA), and the PAM sequence, framing their functions within the design and application of CRISPR-based biosensors for food microbiology.
Molecular Components and Their Mechanisms
crRNA and sgRNA: The Guidance System
The guide RNA is the component that confers specificity to the CRISPR-Cas system. In its natural form in prokaryotes, the guidance system consists of two separate RNA molecules: the CRISPR RNA (crRNA) and the trans-activating CRISPR RNA (tracrRNA) [27]. The crRNA is a short, custom RNA sequence, typically 17–20 nucleotides in length, that is complementary to the target DNA sequence. It is responsible for homology-based recognition and binding to the target site [28] [27]. The tracrRNA, in contrast, serves as a binding scaffold for the Cas nuclease, facilitating the formation of the functional complex [28].
For most laboratory and biosensing applications, these two molecules are combined into a single synthetic molecule known as the single guide RNA (sgRNA). The sgRNA is an engineered RNA molecule that fuses the target-specific crRNA sequence to the structural tracrRNA sequence via a synthetic linker loop [28]. This chimeric design simplifies the system to two main components—the sgRNA and the Cas protein—which is one of the key reasons for CRISPR's widespread adoption. The term "gRNA" is often used generically to refer to all CRISPR guide RNAs, though "sgRNA" specifically denotes this single-molecule format [28].
Table 1: Comparison of CRISPR Guide RNA Components
| Component | Composition | Primary Function | Key Characteristics |
|---|---|---|---|
| crRNA | Short ~20 nt sequence | Target DNA recognition via complementary base pairing | Defines the target locus; customizable for each application [28] [27] |
| tracrRNA | Long, non-coding RNA | Binds Cas nuclease; forms the ribonucleoprotein complex | Provides structural scaffold; constant across different targets [28] [27] |
| sgRNA | Fusion of crRNA + tracrRNA | Combines target recognition and Cas protein binding | Simplified, single-molecule format; most common in research [28] |
PAM Sequence: The Binding Signal
The Protospacer Adjacent Motif (PAM) is a short, specific DNA sequence (typically 2–6 base pairs) located immediately adjacent to the target DNA sequence on the non-complementary strand [9] [27]. Its primary role is to serve as a binding and initiation signal for the Cas nuclease. The Cas protein first scans the DNA for the PAM sequence; only upon recognizing a valid PAM will it unwind the downstream DNA and allow the sgRNA to attempt hybridization with the target strand [9]. This mechanism is crucial for distinguishing between self and non-self DNA in bacterial immunity, preventing the CRISPR system from attacking the bacterium's own genome.
The sequence of the PAM is strictly dependent on the specific Cas nuclease used. For the most common nuclease, SpCas9 from Streptococcus pyogenes, the PAM sequence is 5'-NGG-3', where "N" can be any nucleotide base [9] [28] [27]. This requirement dictates that any genomic target for SpCas9 must have a "GG" dinucleotide immediately following the 20-nucleotide target sequence. It is critical to note that the PAM sequence itself is not part of the sgRNA and is not included in the guide RNA design [28].
Integrated Mechanism of Target Recognition
The process of target recognition is a coordinated, multi-step mechanism. First, the Cas nuclease, in complex with the sgRNA, scans the DNA for a valid PAM sequence. Once a PAM is located, the Cas protein partially unwinds the DNA duplex. The seed sequence—the 8–10 bases at the 3' end of the sgRNA's spacer region—then initiates annealing to the target DNA [9]. If perfect complementarity is achieved in the seed region, annealing continues along the entire spacer sequence. A conformational change in the Cas protein activates its nuclease domains, leading to a double-strand break in the target DNA approximately 3–4 nucleotides upstream of the PAM sequence [9] [27]. For biosensing applications, particularly those using deactivated Cas proteins (dCas9), the nuclease activity is disabled, but the highly specific binding is retained, allowing for the detection and localization of target sequences without cleavage [18] [29].
Application in Microbial Detection: Protocols and Workflows
The high specificity of the crRNA/sgRNA and PAM interaction makes CRISPR-Cas systems ideal for developing biosensors to detect microbial contaminants or to monitor specific strains in food fermentation. The following protocol outlines the workflow for designing and validating a CRISPR-based detection assay for a target bacterial gene.
Protocol: Development of a CRISPR Biosensor for Bacterial Pathogen Detection
Objective: To detect the presence of a specific pathogenic bacterial strain (e.g., Salmonella spp.) in a food sample by targeting a unique genomic sequence with a CRISPR-Cas12a/dCas9-based biosensor.
Principle: A catalytically inactive dCas9 or a reporter-activating Cas12a is programmed with an sgRNA specific to a pathogen gene. Upon binding, a detectable signal (e.g., fluorescence) is generated, confirming the presence of the target microbe [18] [29].
Step 1: Target Gene and sgRNA Design
- Identify Target Gene: Select a unique genomic sequence specific to the pathogen of interest (e.g., an invasion gene in Salmonella). Verify its uniqueness via BLAST against non-target microbial genomes.
- Design sgRNA Spacer Sequence:
- Use sgRNA design software (e.g., CHOPCHOP, Synthego design tool) [28].
- Input the target gene sequence and select the appropriate Cas nuclease (e.g., SpCas9, Cas12a).
- The software will output potential sgRNA spacer sequences (17-23 nt) adjacent to valid PAMs.
- For SpCas9: PAM is 5'-NGG-3', located 3' of the target sequence.
- For Cas12a: PAM is 5'-TTTV-3', located 5' of the target sequence [9] [29].
- Select Optimal sgRNA: Prioritize guides with:
Step 2: sgRNA Synthesis and Complex Formation
- Synthesize sgRNA: Use chemical synthesis for high purity and consistency [28]. Alternatively, employ in vitro transcription (IVT) from a DNA template.
- Form Ribonucleoprotein (RNP): Pre-complex the purified sgRNA with the Cas protein (e.g., dCas9 or Cas12a) in a molar ratio of ~1:1 to 1:2 (Cas:sgRNA) in a suitable buffer. Incubate at 25°C for 10-20 minutes to form the active RNP complex [9].
Step 3: Assay Assembly and Signal Detection
- Sample Preparation: Lyse the food sample (e.g., 1g of meat or produce homogenate) to release microbial DNA. Use a simple heat lysis or a commercial DNA extraction kit.
- Run Detection Assay:
- For a dCas9-based fluorescent system, the RNP may be fused to a fluorescent protein (e.g., GFP). Binding to the target DNA will localize fluorescence, detectable via microscopy or a plate reader [29].
- For a Cas12a-based system, the assay mixture includes:
- Prepared RNP (Cas12a + specific sgRNA).
- Extracted sample DNA.
- A fluorescent reporter probe (e.g., a ssDNA molecule with a fluorophore and quencher).
- Cas12a, upon binding to its target DNA, exhibits collateral cleavage activity, non-specifically cutting the reporter probe and generating a fluorescent signal [29].
- Incubate and Measure: Incubate the reaction at 37°C for 30-60 minutes. Measure fluorescence in real-time or at end-point. A significant increase in fluorescence compared to a negative control confirms detection of the target pathogen.
Step 4: Validation and Quantification
- Specificity Test: Assay against DNA from non-target bacteria to confirm no cross-reactivity.
- Sensitivity (LOD) Determination: Perform the assay with a serial dilution of the target pathogen's DNA to establish the limit of detection.
The Scientist's Toolkit: Essential Reagents for CRISPR Biosensing
Table 2: Key Research Reagent Solutions for CRISPR-based Microbial Detection
| Reagent / Material | Function | Application Notes |
|---|---|---|
| Cas Nuclease (dCas9, Cas12a) | The core enzyme that binds or cleaves DNA upon sgRNA guidance. | dCas9 allows binding without cutting for imaging. Cas12a allows signal amplification via collateral cleavage for sensitive detection [29]. |
| Synthetic sgRNA | The targeting component that defines specificity. | High-purity, synthetic sgRNA ensures consistent performance and high editing efficiency, crucial for assay reproducibility [28]. |
| PAM-containing Target DNA | The DNA sequence from the microbe to be detected. | The target must be adjacent to the correct PAM for the Cas nuclease used (e.g., NGG for SpCas9) [9] [27]. |
| Fluorescent Reporter Probes | Generates a measurable signal upon target recognition. | For Cas12a, a quenched ssDNA probe is cleaved, producing fluorescence. For dCas9, a fused fluorescent protein (e.g., GFP) can be used [29]. |
| sgRNA Design Software | In silico tool for selecting optimal sgRNA sequences. | Tools like CHOPCHOP or Synthego's platform help minimize off-target effects and maximize on-target efficiency [28]. |
The precision of CRISPR-based targeting is governed by the synergistic relationship between the sgRNA (and its constituent crRNA) and the PAM sequence. The programmable nature of the sgRNA provides unparalleled flexibility, allowing researchers to redirect the Cas nuclease to virtually any genomic locus, provided a PAM sequence is nearby. In the specific field of food fermentation research, this translates to the ability to design highly specific biosensors for pathogens like E. coli O157:H7 or Salmonella spp., enabling detection in as little as 20 minutes [18]. As engineering advances yield Cas proteins with altered PAM specificities and enhanced fidelity, the versatility and accuracy of these systems will only increase [9]. A deep understanding of these core components—the crRNA/sgRNA for guidance and the PAM for initial recognition—is therefore foundational to harnessing the full power of CRISPR technology for ensuring food safety and quality.
Food fermentation is a complex biochemical process driven by dynamic microbial ecosystems comprising bacteria, molds, yeasts, and actinomycetes [30]. These microorganisms engage in intricate interactions—including mutualism, commensalism, amensalism, and competition—that ultimately determine the safety, nutritional profile, and sensory characteristics of fermented products [30]. The stability of these microbial communities is influenced by multiple external factors, including raw material variations, environmental conditions (temperature, pH, nutrient composition), and equipment hygiene [30]. This inherent variability presents significant challenges for achieving standardized, high-quality fermented products and necessitates advanced monitoring solutions.
Traditional detection methods, including culture-based techniques, polymerase chain reaction (PCR), and microscopy, while effective, are time-intensive, often requiring several days to yield results [18]. This delay amplifies risks in modern food production, where contaminated products can rapidly enter distribution networks [18]. Within fermentation ecosystems, lactic acid bacteria (LAB) and yeasts frequently demonstrate mutualistic relationships; for example, in grape juice fermentation, yeast provides LAB with amino acids like glutamine, while LAB supply yeast with usable carbon sources, significantly enhancing flavor compound accumulation [30]. Conversely, spoilage organisms and pathogens such as Listeria spp. and Escherichia coli pose significant health risks if undetected [18]. The emergence of CRISPR-based biosensors offers a transformative approach to addressing these monitoring challenges, providing the rapid, specific, and sensitive detection capabilities essential for managing the dynamic microbial ecology of food fermentation [5].
Current Monitoring Challenges in Fermentation Processes
Limitations of Conventional Detection Methods
Conventional pathogen detection methodologies face significant constraints in fermentation monitoring applications. Culture-based techniques, while considered the "gold standard," are time-consuming, labor-intensive, and have low sensitivity, making them unsuitable for rapid response during outbreaks of foodborne disease [6]. Molecular methods such as PCR, while faster than culture methods, require sophisticated infrastructure, expensive equipment, and trained personnel, confining them to central laboratories and limiting their utility in production and processing environments [31] [6]. Furthermore, PCR is susceptible to non-specific amplification that may lead to false-positive results [6]. Immunoassays like ELISA offer simple operation and short detection times but suffer from lower accuracy and sensitivity compared to nucleic acid-based methods [6].
Challenges in Complex Food Matrices
Fermented food matrices present particular challenges for detection technologies. These systems often contain inhibitors such as fats, proteins, and carbohydrates that can interfere with nucleic acid extraction, amplification, and detection processes [5]. These components may reduce the efficiency of enzyme activities or mask target sequences, leading to decreased sensitivity and potential false negatives [5]. Additionally, the dense and diverse microbial communities in fermented foods can create background interference that complicates specific pathogen detection. For starter cultures, the need to monitor viability and metabolic activity in real-time adds another layer of complexity, as traditional methods cannot easily distinguish between live and dead cells or provide insights into microbial functionality [18].
Table 1: Comparison of Pathogen Detection Methods in Food Fermentation
| Method Type | Time to Result | Key Limitations | Sensitivity | Suitable for Real-Time Monitoring |
|---|---|---|---|---|
| Culture-Based | Several days | Labor-intensive, low sensitivity | Moderate | No |
| PCR/qPCR | Hours to 1 day | Requires lab equipment, complex sample prep | High | No |
| ELISA | Several hours | Lower accuracy/sensitivity | Moderate | No |
| Biosensors (General) | Minutes to hours | Matrix interference possible | Moderate to High | Limited |
| CRISPR-Based Biosensors | 20 minutes to 2 hours | Optimization for complex matrices needed | Very High | Yes |
CRISPR-Based Biosensors: Fundamental Principles
CRISPR-Cas System Mechanisms
The CRISPR-Cas system, originally discovered as an adaptive immune mechanism in bacteria and archaea, has emerged as a powerful tool for molecular diagnostics [32] [33]. These systems are categorized into two classes: Class 1 (multi-protein effector complexes) and Class 2 (single effector proteins) [33]. For diagnostic applications, Class II systems are predominantly employed due to their simplicity and programmability [33]. Key effector proteins include:
- Cas9: The first Cas protein applied in nucleic acid detection, Cas9 recognizes and cleaves double-stranded DNA (dsDNA) containing a protospacer adjacent motif (PAM) sequence. While useful, it possesses only cis-cleavage activity [33].
- Cas12: A Type V effector that recognizes dsDNA or ssDNA with a PAM sequence and exhibits robust trans-cleavage activity, nonspecifically cleaving single-stranded DNA (ssDNA) upon target recognition [6] [33].
- Cas13: A Type VI effector that binds to target single-stranded RNA (ssRNA) and promiscuously cleaves surrounding RNA molecules through its trans-cleavage activity [33].
The trans-cleavage activity of Cas12 and Cas13 proteins forms the foundation for most CRISPR-based diagnostic applications. When these Cas proteins bind to their target nucleic acid sequences guided by CRISPR RNA (crRNA), they undergo conformational changes that activate their collateral cleavage capabilities, indiscriminately degrading nearby reporter molecules [6].
Signal Readout Mechanisms
CRISPR-based biosensors employ various signal readout mechanisms to detect pathogen presence:
- Fluorescence: The most common readout method utilizes reporter molecules consisting of a fluorophore and quencher linked by a nucleic acid backbone. When cleaved by activated Cas proteins, the fluorophore separates from the quencher, generating a fluorescent signal [33].
- Colorimetric: These systems use gold nanoparticles (AuNPs) or other color-changing reagents that aggregate or disperse based on Cas-mediated cleavage events, producing visible color changes detectable by eye or simple readers [32] [33].
- Lateral Flow: Similar to pregnancy tests, these strips detect cleaved reporter molecules, providing a simple, equipment-free readout suitable for field use [31].
Table 2: CRISPR-Cas Systems for Pathogen Detection in Food Safety
| CRISPR System | Target Type | PAM Sequence | Trans-Cleavage Activity | Key Applications |
|---|---|---|---|---|
| Cas9 | dsDNA | 5'-NGG-3' | None | Nucleic acid detection with PAMmers |
| Cas12a | dsDNA, ssDNA | 5'-TTN-3' | ssDNA | DETECTR for bacterial pathogens |
| Cas13a | ssRNA | None | ssRNA | SHERLOCK for viral pathogens |
| Cas12f | ssDNA | None | ssDNA | Compact system for small targets |
Application Notes: Implementing CRISPR Biosensors in Food Fermentation
Pathogen Detection in Complex Fermentation Matrices
CRISPR-based biosensors have demonstrated remarkable capabilities for detecting foodborne pathogens in various fermentation matrices. The technology enables rapid identification of bacterial contaminants like Salmonella, Escherichia coli, and Listeria monocytogenes with sensitivity comparable to or exceeding traditional methods [5]. For example, Cas12-based systems have detected E. coli O157:H7 in as little as 20 minutes using microelectrode arrays [18], while Cas13-based systems have identified Salmonella spp. through nucleic acid-based sensors [18].
Implementation in fermented products requires matrix-specific optimization:
- Dairy Products: For yogurt and cheese, sample preparation must account for high fat and protein content that may inhibit amplification. Pre-treatment with proteinases and dilution buffers improves detection efficiency.
- Fermented Meats: In sausage products, salt content and spices may interfere; incorporating chelating agents in extraction buffers mitigates these effects.
- Plant-Based Ferments: For kimchi and sauerkraut, high acidity requires pH neutralization before nucleic acid extraction to preserve enzyme activity.
The integration of CRISPR with isothermal amplification methods like Recombinase Polymerase Amplification (RPA) or Loop-Mediated Isothermal Amplification (LAMP) enhances sensitivity without requiring thermal cyclers, making these systems suitable for field-deployable diagnostics [31]. This combination has enabled detection limits as low as single copy numbers of target pathogens in some applications [5].
Starter Culture Performance Monitoring
Beyond pathogen detection, CRISPR biosensors offer innovative approaches for monitoring starter culture viability and functionality. Traditional methods for tracking starter cultures like Lactobacillus spp. and Streptococcus spp. require days, while isothermal microcalorimetry detection of Lactobacillus plantarum was achieved in 4.7–18.6 hours [18]. CRISPR-based approaches can potentially reduce this timeframe to hours or minutes by targeting species-specific genetic markers.
Key applications for starter culture monitoring include:
- Fermentation Progress Tracking: Quantifying dominant microbial populations throughout fermentation to optimize process parameters.
- Culture Purity Verification: Ensuring starter cultures remain free of phage contamination or competitor strains.
- Metabolic Activity Assessment: Monitoring expression of key genes involved in flavor compound production.
The programmability of CRISPR systems enables multiplexed detection of multiple starter culture strains simultaneously, providing comprehensive insights into population dynamics [31]. This capability is particularly valuable for complex fermented products requiring specific microbial consortia, such as kefir or sourdough, where balanced microbial composition is essential for product quality [30].
Experimental Protocols
Protocol 1: Detection of Listeria monocytogenes in Fermented Dairy Products
Principle: This protocol utilizes Cas12a's trans-cleavage activity to detect Listeria monocytogenes contamination in yogurt and cheese. The assay combines RPA pre-amplification with CRISPR-Cas12a detection and fluorescence readout.
Materials and Reagents:
- Cas12a Enzyme: Purified LbCas12a or AsCas12a protein
- crRNA: Designed against L. monocytogenes hlyA gene sequence
- RPA Kit: Basic RPA kit with lyophilized pellets
- Fluorescent Reporter: FAM-TTATT-BHQ1 ssDNA reporter
- Sample Preparation: Lysis buffer (Tris-EDTA with lysozyme and proteinase K)
- Equipment: Fluorescence reader or lateral flow strip reader
Procedure:
- Sample Preparation:
- Homogenize 1g of fermented dairy sample with 5mL of lysis buffer
- Incubate at 55°C for 15 minutes with vortexing every 5 minutes
- Centrifuge at 12,000 × g for 2 minutes and collect supernatant
RPA Pre-amplification:
- Prepare RPA reaction mix according to manufacturer's instructions
- Add 2μL of sample supernatant to RPA reaction tube
- Incubate at 37°C for 20 minutes
CRISPR-Cas12a Detection:
- Prepare Cas12a detection mix:
- 200nM Cas12a
- 400nM hlyA-specific crRNA
- 500nM fluorescent reporter
- 1× NEBuffer 2.1
- Combine 5μL of RPA product with 15μL of Cas12a detection mix
- Incubate at 37°C for 10-15 minutes
- Prepare Cas12a detection mix:
Signal Detection:
- Measure fluorescence at 485nm excitation/520nm emission
- Alternatively, apply reaction to lateral flow strip and interpret after 5 minutes
Interpretation: Fluorescence signal ≥3× background or visible test line on lateral flow strip indicates L. monocytogenes contamination.
Protocol 2: Multiplex Monitoring of Starter Cultures in Sourdough Fermentation
Principle: This protocol employs Cas13 and Cas12f for simultaneous detection of Lactobacillus sanfranciscensis and Candida milleri, key microorganisms in sourdough fermentation.
Materials and Reagents:
- Cas Proteins: LwaCas13a (for RNA detection) and Cas12f (for DNA detection)
- crRNAs: Species-specific crRNAs for L. sanfranciscensis (targeting 16S rRNA) and C. milleri (targeting ITS region)
- Reporters: FAM-UUUU-BHQ1 for Cas13; HEX-TTATT-Iowa Black for Cas12f
- Extraction Kit: Dual DNA/RNA extraction kit
- Equipment: Real-time fluorometer or plate reader
Procedure:
- Nucleic Acid Co-extraction:
- Extract total nucleic acids from 0.5g sourdough sample using commercial kit
- Elute in 50μL nuclease-free water
- Quantify DNA/RNA concentration by spectrophotometry
CRISPR Detection Setup:
- Prepare master mix containing:
- 100nM LwaCas13a
- 150nM Cas12f
- 300nM each crRNA
- 500nM each fluorescent reporter
- 1× reaction buffer
- Distribute 18μL aliquots into reaction tubes
- Add 2μL nucleic acid extract to each reaction
- Prepare master mix containing:
Incubation and Measurement:
- Incubate reactions at 37°C with continuous fluorescence monitoring
- Read FAM channel (Cas13) and HEX channel (Cas12f) every minute for 30 minutes
Data Analysis:
- Calculate threshold time (Tt) for each fluorescence channel
- Generate standard curves using known concentrations of pure cultures
- Quantify target microorganisms in samples using standard curves
Interpretation: The ratio of L. sanfranciscensis to C. milleri should approximate 100:1 for optimal sourdough fermentation quality. Significant deviations indicate fermentation abnormalities requiring intervention.
The Scientist's Toolkit: Essential Research Reagents
Table 3: Essential Reagents for CRISPR-Based Monitoring in Food Fermentation
| Reagent Category | Specific Examples | Function | Application Notes |
|---|---|---|---|
| Cas Proteins | LbCas12a, AsCas12a, LwaCas13a | Target recognition and collateral cleavage | Cas12a for DNA targets, Cas13a for RNA targets |
| Guide RNAs | crRNAs targeting pathogen-specific genes | Programmable target recognition | Design against unique genomic regions; avoid conserved sequences |
| Amplification Reagents | RPA pellets, LAMP kits | Nucleic acid pre-amplification | Enables detection of low pathogen concentrations |
| Reporters | FAM/TTATT/BHQ1, FAM/UUUU/BHQ1 | Signal generation upon target detection | ssDNA reporters for Cas12; ssRNA for Cas13 |
| Sample Preparation | Lysozyme, proteinase K, lysis buffers | Nucleic acid release and purification | Critical for complex food matrices |
| Readout Systems | Fluorescence readers, lateral flow strips | Result visualization | Choice depends on required sensitivity and portability |
CRISPR-based biosensors represent a paradigm shift in monitoring microbial ecology during food fermentation. By providing rapid, specific, and sensitive detection of pathogens and starter cultures, these technologies address critical gaps in current food safety and quality control systems [5]. The integration of CRISPR diagnostics with isothermal amplification techniques and portable readout systems creates powerful tools for decentralized testing, enabling real-time decision-making in production environments [31].
Future developments in CRISPR-based monitoring will likely focus on several key areas:
- Multiplexing Capabilities: Engineering systems capable of simultaneously detecting multiple pathogens, spoilage organisms, and starter cultures in a single reaction [5].
- Point-of-Use Devices: Creating integrated, equipment-free platforms for routine monitoring in production facilities [31].
- Quantitative Detection: Enhancing systems to provide quantitative data on microbial loads rather than mere presence/absence results [6].
- Non-Nucleic Acid Targets: Expanding CRISPR applications to detect small molecules, toxins, and other non-nucleic acid biomarkers relevant to fermentation quality [32] [33].
The transformative potential of CRISPR technology in food fermentation aligns with broader initiatives such as One Health, connecting food safety with public health outcomes [5]. As these biosensors become more accessible and standardized, they promise to revolutionize how we monitor, understand, and control microbial ecosystems in fermented foods, ultimately leading to safer, higher-quality products for consumers worldwide.
Building the Sensor: Methodologies and Direct Applications in Fermentation Systems
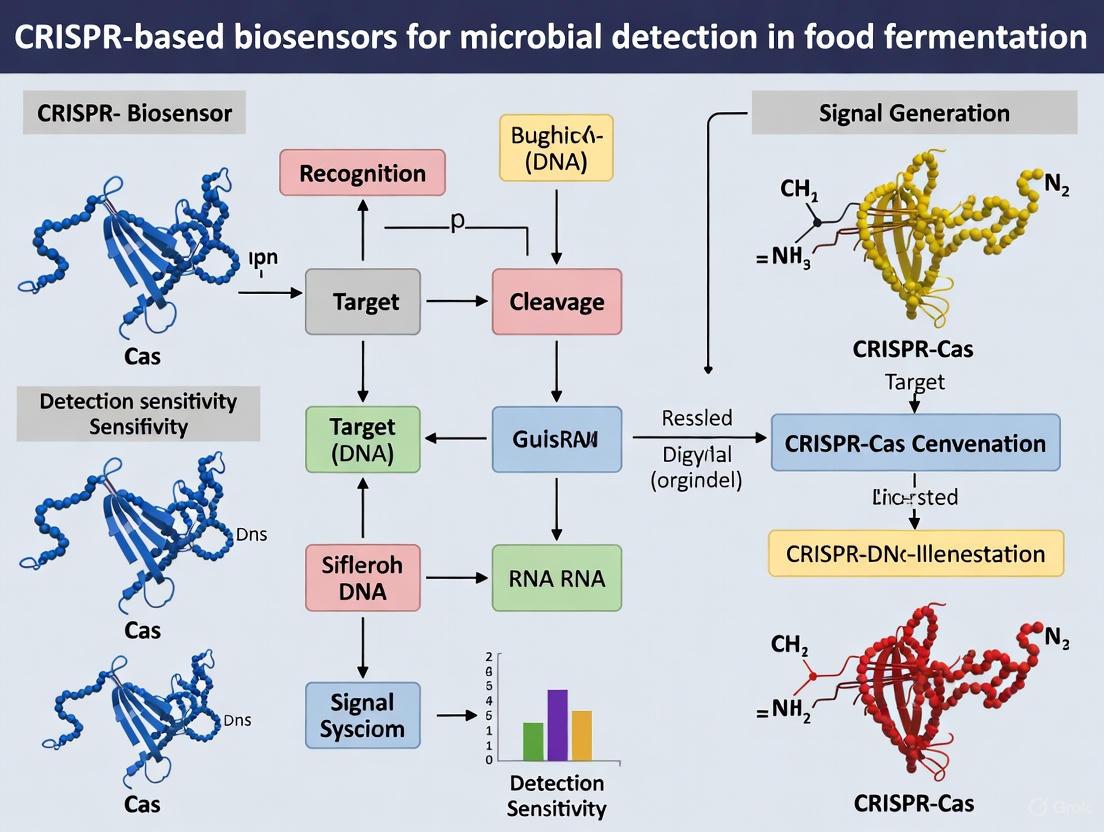
The SHERLOCK (Specific High-sensitivity Enzymatic Reporter unLOCKing) and DETECTR (DNA Endonuclease Targeted CRISPR Trans Reporter) systems represent transformative CRISPR-based diagnostic platforms that have redefined the standards for pathogen detection. These technologies leverage the collateral cleavage activity of different Cas enzymes—Cas13 for RNA targeting in SHERLOCK and Cas12 for DNA targeting in DETECTR—to achieve attomolar sensitivity and single-base specificity [5] [34]. Within food fermentation research, these systems offer unprecedented capabilities for monitoring microbial communities, detecting contaminating pathogens, and ensuring the quality and safety of fermented products in real-time [5] [35]. Their rapid detection timeframe of 30-60 minutes and compatibility with portable formats make them particularly valuable for quality control at production facilities, reducing reliance on centralized laboratories and enabling proactive intervention in fermentation processes [36] [37].
The SHERLOCK (Cas13) Platform
Principles and Mechanisms
The SHERLOCK system utilizes Cas13a, an RNA-guided RNA nuclease belonging to the type VI CRISPR-Cas system [38]. Upon recognition and cis-cleavage of its target single-stranded RNA (ssRNA) sequence, Cas13a exhibits promiscuous trans-cleavage activity, indiscriminately degrading surrounding non-target ssRNA molecules [36] [39]. This collateral activity is harnessed for diagnostics by introducing engineered reporter RNAs—typically labeled with a fluorophore and a quencher. When intact, the reporter molecule emits no signal, but upon cleavage by the activated Cas13a, the fluorophore is separated from the quencher, generating a fluorescent signal that confirms target detection [38] [39]. The requirement for both target recognition and a specific protospacer flanking sequence (PFS) ensures high specificity, enabling SHERLOCK to distinguish between even closely related microbial strains [38].
Applications in Food Fermentation Research
SHERLOCK is ideally suited for applications requiring RNA detection. In food fermentation, this includes:
- Direct Viral Pathogen Detection: Rapid identification of RNA viruses that can compromise fermentation starters or pose safety risks, such as Norovirus [38].
- Microbial Metabolism Monitoring: Quantifying specific mRNA biomarkers to monitor the metabolic activity of starter cultures (e.g., lactic acid bacteria) during fermentation [39].
- Fungal Contaminant Screening: Detecting mycotoxin-producing fungi by targeting their messenger RNA, allowing for early intervention in fermentations involving grains or other susceptible substrates [5].
The following diagram illustrates the core mechanism of the SHERLOCK platform:
Detailed SHERLOCK Protocol
Objective: To detect a specific bacterial RNA marker (e.g., a virulence gene from Listeria monocytogenes) in a fermented food sample (e.g., cheese).
Workflow:
- Nucleic Acid Extraction (15 min): Extract total RNA from a 1g food sample using a commercial kit. For complex matrices, include an additional inhibitor removal step.
- Reverse Transcription & Isothermal Amplification (25 min at 41°C):
- Prepare a 25 µL RPA reaction mix containing:
- 5 µL of extracted RNA.
- Primers (2 µM each) targeting the L. monocytogenes hlyA gene.
- Reverse transcriptase (for integrated RT-RPA).
- RPA rehydration buffer and enzyme pellet.
- Incubate the mixture at 41°C for 25 minutes. This step simultaneously reverse transcribes RNA to cDNA and exponentially amplifies the target DNA.
- Prepare a 25 µL RPA reaction mix containing:
- CRISPR-Cas13 Detection (10 min at 37°C):
- Prepare a 10 µL Cas13 detection mix containing:
- LbaCas13a or LwaCas13a (50 nM).
- Target-specific crRNA (50 nM).
- Fluorescent RNA Reporter (100 nM).
- Nuclease-free buffer.
- Transfer 2 µL of the RPA amplification product into the detection mix.
- Incubate at 37°C for 10 minutes and measure fluorescence using a portable fluorometer or a plate reader. A positive result is indicated by a significant increase in fluorescence over a no-template control.
- Prepare a 10 µL Cas13 detection mix containing:
The DETECTR (Cas12) Platform
Principles and Mechanisms
The DETECTR system employs Cas12a (e.g., LbCas12a or AsCas12a), a DNA-guided DNA nuclease from the type V CRISPR-Cas system [37] [40]. Similar to Cas13, Cas12a possesses collateral trans-cleavage activity; however, it is activated upon binding to a complementary double-stranded DNA (dsDNA) target and non-specifically cleaves surrounding single-stranded DNA (ssDNA) molecules [37] [40]. A key distinction is its requirement for a short Protospacer Adjacent Motif (PAM) sequence (5'-TTTV-3' for LbCas12a) immediately adjacent to the target DNA for initial recognition and activation [37]. For detection, a fluorescently labeled ssDNA reporter is used. The activated Cas12a cleaves this reporter, producing a detectable signal that confirms the presence of the target DNA sequence [40].
Applications in Food Fermentation Research
DETECTR excels in DNA-based applications critical to fermentation science:
- Bacterial Pathogen Identification: Sensitive detection of DNA from bacterial contaminants like Salmonella spp., E. coli O157:H7, and Listeria monocytogenes in raw ingredients or during fermentation [5] [37].
- Strain Verification: Confirming the identity and purity of proprietary bacterial or yeast starter cultures by targeting unique genomic signatures [35].
- Spoilage Microbe Monitoring: Tracking the emergence of spoilage bacteria (e.g., Acetobacter or certain Lactobacillus strains) that can negatively impact product quality [5].
The following diagram illustrates the core mechanism of the DETECTR platform:
Detailed DETECTR Protocol
Objective: To detect the genomic DNA of Salmonella enterica in a fermented sausage sample.
Workflow:
- DNA Extraction (15 min): Extract genomic DNA from a 1g sausage sample using a commercial kit designed for complex food matrices.
- Isothermal Amplification (20 min at 39°C):
- Prepare a 50 µL RPA reaction mix containing:
- 10 µL of extracted DNA.
- Primers (0.3 µM each) targeting the S. enterica invA gene.
- RPA rehydration buffer, magnesium acetate, and enzyme pellet.
- Incubate at 39°C for 20 minutes to amplify the target DNA region.
- Prepare a 50 µL RPA reaction mix containing:
- CRISPR-Cas12 Detection (10 min at 37°C):
- Prepare a 20 µL Cas12 detection mix containing:
- LbCas12a (50 nM).
- Target-specific crRNA (50 nM) designed to bind the amplified invA region with a compatible PAM.
- Fluorescent ssDNA Reporter (100 nM).
- Nuclease-free buffer.
- Transfer 5 µL of the RPA amplification product into the detection mix.
- Incubate at 37°C for 10 minutes. The result can be read via fluorescence or by applying the reaction to a lateral flow dipstick for a visual colorimetric readout [37] [40].
- Prepare a 20 µL Cas12 detection mix containing:
Comparative Performance Analysis
The table below summarizes the key characteristics of the SHERLOCK and DETECTR platforms for easy comparison.
Table 1: Comparative Analysis of SHERLOCK and DETECTR Diagnostic Platforms
| Parameter | SHERLOCK (Cas13) | DETECTR (Cas12) |
|---|---|---|
| CRISPR Effector | Cas13a, Cas13b [38] | Cas12a (Cpf1), Cas12b [37] [40] |
| Native Target | Single-stranded RNA (ssRNA) [36] [38] | Double-stranded DNA (dsDNA) [37] [40] |
| Collateral Activity | Trans-cleavage of ssRNA [39] | Trans-cleavage of ssDNA [37] |
| Key Sequence Motif | Protospacer Flanking Site (PFS) [38] | Protospacer Adjacent Motif (PAM) [37] |
| Typical Sensitivity | Attomolar (aM) [36], <10 copies/µL [36] | Attomolar (aM) [34] |
| Assay Time | 30 - 60 minutes [36] | 20 - 40 minutes [37] [40] |
| Primary Application in Food Fermentation | Detection of RNA viruses, metabolic activity monitoring, fungal screening [5] [39] | Detection of bacterial pathogens, strain verification, spoilage monitoring [5] [37] |
| Common Readout Methods | Fluorescence, Lateral Flow Strips [36] [38] | Fluorescence, Lateral Flow Strips [37] [40] |
The Scientist's Toolkit: Essential Research Reagents
Successful implementation of SHERLOCK and DETECTR assays requires a core set of reagents and tools.
Table 2: Essential Reagents and Materials for CRISPR-Based Diagnostics
| Reagent / Material | Function | Example Specifications |
|---|---|---|
| Purified Cas Enzyme | The core effector protein that performs targeted recognition and collateral cleavage. | LbaCas13a, LwaCas13a for SHERLOCK; LbCas12a for DETECTR [38] [37]. |
| Synthetic crRNA | Guides the Cas enzyme to the specific target nucleic acid sequence. | Custom-designed, target-specific crRNA; ~20 nt spacer for Cas13a [38], requires PAM-compatible design for Cas12a [37]. |
| Fluorescent Reporter | Signal-generating molecule cleaved during collateral activity. | ssRNA reporter (e.g., 5'-FAM/UUU/3'-IABkFQ) for SHERLOCK; ssDNA reporter (e.g., 5'-6-FAM/TTATT/3'-BHQ1) for DETECTR [38] [40]. |
| Isothermal Amplification Kit | Pre-amplifies the target to achieve high sensitivity. | RPA (TwistAmp) or LAMP kits [36] [37]. |
| Nucleic Acid Extraction Kit | Isolates high-quality DNA/RNA from complex food matrices. | Kits with inhibitor removal technology for fermented foods [5]. |
| Lateral Flow Strips | For simple, equipment-free visual readout. | Streptavidin control line, anti-FAM test line; compatible with biotin- and FAM-labeled reporters [40]. |
The SHERLOCK and DETECTR platforms provide robust, rapid, and highly specific diagnostic tools that are ideally suited for addressing complex challenges in food fermentation research. By enabling on-site detection of pathogens, verification of starter cultures, and monitoring of microbial dynamics, these CRISPR-based biosensors empower researchers and producers to enhance the safety, quality, and consistency of fermented products. As these technologies continue to evolve with integration into portable devices and AI-driven analysis, their role in building a more resilient and sustainable food system is poised to expand significantly [5] [26].
The integration of Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR) technology with isothermal amplification techniques, particularly Recombinase Polymerase Amplification (RPA) and Loop-Mediated Isothermal Amplification (LAMP), represents a transformative advancement in molecular diagnostics for food fermentation research. This combination leverages the rapid, equipment-free nucleic acid amplification of RPA and LAMP with the precise targeting and collateral activity of CRISPR-Cas systems, enabling unprecedented sensitivity and specificity in detecting microbial contaminants and starter cultures. These platforms achieve detection limits as low as 1-10 copies per μL and diagnostic specificity ranging from 95% to 100%, making them superior to traditional culture methods and PCR-based assays for point-of-need testing. This application note provides detailed protocols and methodological frameworks for implementing RPA-CRISPR and LAMP-CRISPR biosensors in food fermentation quality control, supporting researchers in the rapid detection of pathogens such as Staphylococcus aureus, Salmonella species, and spoilage microorganisms in complex fermented matrices.
In food fermentation research, ensuring microbial purity and monitoring starter culture integrity are critical for product quality and safety. Traditional detection methods, including culture-based techniques and polymerase chain reaction (PCR), present significant limitations for in-process monitoring, including time-consuming procedures, extensive labor requirements, and dependency on sophisticated laboratory equipment. The emergence of isothermal amplification techniques, particularly RPA and LAMP, has revolutionized molecular diagnostics by enabling rapid nucleic acid amplification at constant temperatures without thermal cycling.
RPA operates at 37-42°C through the synergistic activity of recombinase proteins, single-stranded DNA-binding proteins, and strand-displacing polymerases, achieving detectable amplification within 10-30 minutes [41]. LAMP employs 4-6 primers targeting 6-8 distinct regions and a strand-displacing Bst DNA polymerase active at 60-65°C, typically completing amplification within 15-60 minutes [42]. Both techniques offer significant advantages for resource-limited settings and point-of-care applications, though they occasionally suffer from non-specific amplification and false-positive results.
The integration of these methods with CRISPR-Cas systems has created a new paradigm in detection technology. CRISPR-Cas12a, upon recognition of its target DNA sequence, exhibits both specific cis-cleavage activity and non-specific trans-cleavage activity, enabling highly specific detection through collateral cleavage of reporter molecules [41]. When combined with RPA or LAMP pre-amplification, CRISPR-Cas systems provide a secondary verification step that enhances overall detection specificity and sensitivity, making these integrated platforms particularly valuable for detecting low-abundance microbial targets in complex food matrices such as fermented products.
Technical Comparison of RPA and LAMP Platforms
The selection of an appropriate isothermal amplification technique depends on multiple factors, including required sensitivity, reaction conditions, and detection throughput. The table below provides a comprehensive comparison of RPA and LAMP methodologies for integration with CRISPR-based detection systems.
Table 1: Technical Comparison of RPA and LAMP Platforms for CRISPR Integration
| Parameter | Recombinase Polymerase Amplification (RPA) | Loop-Mediated Isothermal Amplification (LAMP) |
|---|---|---|
| Reaction Temperature | 37-42°C | 60-65°C |
| Reaction Time | 10-30 minutes | 15-60 minutes |
| Sensitivity | 1-100 copies/μL [41] | 0.01-100 copies/μL [43] |
| Primer Requirements | 2 primers (30-35 bp) | 4-6 primers (15-25 bp) [42] |
| Key Enzymes | Recombinase, SSB, strand-displacing polymerase | Bst DNA polymerase with strand displacement activity |
| Equipment Cost | Low | Low to medium |
| Advantages | Rapid, lower temperature, simple primer design | High sensitivity, robust amplification |
| Disadvantages | Primer design critical, limited throughput | Complex primer design, non-specific amplification risk |
| Optimal CRISPR Partner | Cas12a (37°C compatibility) | Cas12b (thermostable variants) |
Beyond these fundamental characteristics, method selection should consider additional practical factors. RPA's lower operating temperature (37-42°C) makes it compatible with portable devices and field applications, while LAMP's higher temperature requirement (60-65°C) provides inherent protection against non-specific amplification but may limit device portability. For sensitivity-critical applications, LAMP generally offers superior detection limits, with demonstrated capability to detect as low as 0.01 ng/μL genomic DNA, approximately 10 times more sensitive than real-time PCR and 100 times more sensitive than conventional PCR [43]. However, RPA-CRISPR combinations have achieved impressive results, with documented detection of Salmonella typhimurium at concentrations as low as 3 CFU/mL [44].
Experimental Protocols
RPA-CRISPR/Cas12a Detection Protocol
This protocol details a standardized approach for detecting microbial targets in food fermentation samples using RPA pre-amplification followed by CRISPR-Cas12a-mediated detection, adaptable for various bacterial and fungal targets relevant to fermentation processes.
Sample Preparation and Nucleic Acid Extraction
- Sample Collection: Aseptically collect 10g of fermenting material and homogenize with 90mL of sterile phosphate-buffered saline (PBS).
- Microbial Enrichment: Incubate homogenate at appropriate temperature (e.g., 37°C for bacteria, 30°C for yeast) for 6-8 hours to enrich target microorganisms.
- Nucleic Acid Extraction: Extract genomic DNA using commercial kits or a simplified thermal lysis method (heating at 95°C for 10 minutes in chelating buffer) [45].
- DNA Quantification: Measure DNA concentration using spectrophotometry and dilute to working concentration (typically 1-10 ng/μL) in nuclease-free water.
RPA Pre-amplification Reaction
Table 2: RPA Reaction Setup
| Component | Volume | Final Concentration |
|---|---|---|
| RPA rehydration buffer | 25.4 μL | 1× |
| Forward primer (10 μM) | 1.0 μL | 0.4 μM |
| Reverse primer (10 μM) | 1.0 μL | 0.4 μM |
| Template DNA | 5.0 μL | 1-10 ng/μL |
| Nuclease-free water | 10.6 μL | - |
| Magnesium acetate (280 mM) | 2.5 μL | 14 mM |
Procedure:
- Prepare master mix containing all components except magnesium acetate.
- Aliquot 47.5 μL of master mix into 0.2 mL RPA reaction tubes.
- Initiate amplification by adding 2.5 μL of magnesium acetate solution.
- Incubate at 39°C for 15-20 minutes [45].
- Briefly centrifuge to collect condensation before proceeding to CRISPR detection.
CRISPR-Cas12a Detection
Table 3: CRISPR-Cas12a Reaction Setup
| Component | Volume | Final Concentration |
|---|---|---|
| Cas12a enzyme (10 μM) | 1.0 μL | 100 nM |
| crRNA (10 μM) | 1.0 μL | 100 nM |
| NEBuffer 2.1 | 2.5 μL | 1× |
| ssDNA reporter (10 μM) | 1.0 μL | 1 μM |
| RPA amplicon | 2.0 μL | Undiluted |
| Nuclease-free water | 17.5 μL | - |
Procedure:
- Combine all components in a clean reaction tube.
- Incubate at 37°C for 10-15 minutes.
- Visualize results using blue light transillumination (fluorescence) or lateral flow strips.
LAMP-CRISPR/Cas12b Detection Protocol
This protocol describes the use of LAMP pre-amplification coupled with thermostable CRISPR-Cas12b detection, particularly suitable for targets requiring higher reaction temperatures or enhanced specificity.
LAMP Pre-amplification Reaction
Table 4: LAMP Reaction Setup
| Component | Volume | Final Concentration |
|---|---|---|
| Isothermal amplification buffer | 12.5 μL | 1× |
| LAMP primer mix (FIP/BIP: 4 μM each; F3/B3: 0.5 μM each) | 2.5 μL | Variable |
| Bst DNA polymerase (8 U/μL) | 1.0 μL | 0.32 U/μL |
| MgSO₄ (100 mM) | 1.0 μL | 8 mM |
| Betaine (5 M) | 4.0 μL | 0.8 M |
| dNTPs (10 mM) | 2.0 μL | 1.4 mM |
| Template DNA | 2.0 μL | 1-10 ng/μL |
| Nuclease-free water | 4.0 μL | - |
Procedure:
- Prepare master mix containing all components except template DNA.
- Aliquot 23 μL of master mix into reaction tubes.
- Add 2 μL of template DNA to each reaction.
- Incubate at 65°C for 45-60 minutes [43].
- Heat-inactivate at 80°C for 5 minutes before CRISPR detection.
CRISPR-Cas12b Detection
Table 5: CRISPR-Cas12b Reaction Setup
| Component | Volume | Final Concentration |
|---|---|---|
| Cas12b enzyme (10 μM) | 1.0 μL | 100 nM |
| crRNA (10 μM) | 1.0 μL | 100 nM |
| Thermopol buffer | 2.5 μL | 1× |
| ssDNA reporter (10 μM) | 1.0 μL | 1 μM |
| LAMP amplicon | 2.0 μL | 1:10 dilution |
| Nuclease-free water | 17.5 μL | - |
Procedure:
- Combine all components in a reaction tube.
- Incubate at 55°C for 15 minutes.
- Detect fluorescence using a portable reader or visual inspection.
The Scientist's Toolkit: Essential Research Reagent Solutions
Successful implementation of RPA-CRISPR and LAMP-CRISPR detection platforms requires carefully selected reagents and components. The following table details essential research reagent solutions for establishing these diagnostic workflows.
Table 6: Essential Research Reagent Solutions for RPA/LAMP-CRISPR Integration
| Reagent Category | Specific Examples | Function & Importance | Optimization Notes |
|---|---|---|---|
| Recombinase Enzymes | T4 UvsX recombinase | Binds primers and invades dsDNA to initiate RPA | Maintain fresh stocks; avoid freeze-thaw cycles |
| Strand-Displacing Polymerases | Bst DNA polymerase (LAMP), Sau polymerase (RPA) | Extends primers while displacing downstream strands | Magnesium concentration critical for activity |
| CRISPR Enzymes | LbCas12a, AsCas12a, AaCas12b | Target-specific binding and collateral cleavage activity | Cas12a for lower temps (37°C), Cas12b for higher (55°C) |
| Guide RNA | crRNA with target-specific spacer | Directs Cas protein to complementary nucleic acid sequences | Design spacers 20-25 nt; verify PAM requirements |
| Reporters | FAM-biotin-ssDNA, FQ-labeled reporters (FAM/TAMRA/BHQ1) | Trans-cleavage substrate for signal generation | Quencher selection affects signal-to-noise ratio |
| Primer Design | RPA (30-35 bp), LAMP (FIP/BIP/F3/B3/LF/LB) | Target-specific amplification | Avoid secondary structures; verify specificity |
| Reaction Enhancers | PEG, crowding agents (RPA); Betaine, DMSO (LAMP) | Enhance enzyme stability and reaction efficiency | Concentration optimization required for each target |
Advanced Applications and Methodological Innovations
One-Pot Assay Configurations
Recent advancements have focused on developing one-pot assays that integrate amplification and detection in a single tube, minimizing contamination risk and simplifying operational workflows. Several strategic approaches have emerged:
Physical Separation Methods: Initial approaches used spatial separation of RPA and CRISPR reagents within the same tube (e.g., RPA in tube cap, CRISPR in bottom), requiring manual mixing after amplification [46]. While reducing tube transfers, this method still poses contamination risks during mixing.
Light-Activated Systems: Advanced systems incorporate photocleavable linkers in tailed crRNA designs, maintaining CRISPR system inactivity during RPA amplification until ultraviolet irradiation activates detection [46]. This approach enables true single-tube reactions with minimal cross-contamination, though with slightly reduced sensitivity (LOD: 34.7 CFU/mL vs 6.3 CFU/mL in two-step methods).
Temperature-Phase Separation: Systems utilizing glycerol-based phase separation or thermostable Cas variants enable sequential activation of amplification and detection through temperature modulation without physical manipulation [46].
PAM-Independent Detection Systems
Traditional CRISPR-Cas systems require specific Protospacer Adjacent Motif (PAM) sequences adjacent to target sites, limiting target flexibility. Innovative approaches now circumvent this limitation:
Peptide Nucleic Acid (PNA)-Assisted Systems: PNAs precisely invade double-stranded DNA to form single-stranded regions, enabling Cas12a recognition without PAM requirements. This PNA-assisted self-folding isothermal amplification approach has demonstrated remarkable sensitivity of 3 CFU/mL for Salmonella typhimurium detection [44].
Self-Primer EXPAR Systems: Self-primer exponential amplification reaction (SP-EXPAR) simultaneously generates both double-stranded DNA (with PAM) and single-stranded DNA (without PAM) products, both detectable by CRISPR-Cas12a, enhancing overall detection sensitivity to 7.49 aM for SARS-CoV-2 variants [47]. This approach has relevance for detecting RNA viruses that may contaminate fermentation systems.
Multiplex Detection Capabilities
The modular nature of CRISPR-based detection enables multiplexing for simultaneous identification of multiple microbial targets:
crRNA Cocktails: Combining multiple target-specific crRNAs in a single reaction allows parallel detection of different pathogens or genetic markers.
Differential Reporter Systems: Using spectrally distinct fluorophores (e.g., FAM, TAMRA, Cy5) or different haptens for lateral flow detection enables simultaneous detection of multiple targets in a single reaction.
Performance Assessment and Validation
RPA-CRISPR and LAMP-CRISPR platforms consistently demonstrate exceptional performance characteristics suitable for quality control in food fermentation environments:
Sensitivity and Specificity: In systematic evaluations, these integrated platforms achieved diagnostic sensitivity of 95-100% and specificity of 100% for Mycobacterium tuberculosis detection compared to reference methods like GeneXpert and culture [48]. Similar performance can be anticipated for fermentation-relevant microorganisms with proper optimization.
Limit of Detection (LOD): The exceptional sensitivity of these methods enables detection of extremely low target concentrations. RPA-CRISPR platforms have demonstrated limits of detection as low as 1 copy per μL for tuberculosis detection [48] and 100 fg/μL for fungal pathogens [43], sufficient for detecting low-level contamination in fermentation systems.
Robustness in Complex Matrices: When applied to food matrices including lettuce, milk, salmon, and sauce beef, RPA-CRISPR systems maintained excellent recovery rates (87.1-110.2%) and reproducibility (RSD ≤ 6.22%) [46], indicating reliable performance in complex fermentation matrices containing inhibitors.
Speed and Throughput: Complete analysis from sample to results typically requires 45-90 minutes, significantly faster than culture methods (2-5 days) or conventional PCR with gel electrophoresis (2-3 hours). This rapid turnaround enables near real-time monitoring of fermentation processes.
The precise monitoring of microbial populations is paramount in food fermentation research, ensuring product consistency, safety, and quality. Traditional microbiological methods are often labor-intensive and time-consuming, failing to provide the real-time data necessary for proactive process control. The integration of Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR)-based biosensors has revolutionized this field by offering unparalleled specificity and sensitivity for detecting microbial targets. The efficacy of these biosensors is fundamentally governed by their signal readout strategies. This article details the core principles, protocols, and applications of four primary readout modalities—fluorescence, colorimetric, lateral flow, and electrochemical—providing a structured guide for their implementation in food fermentation research.
Comparative Analysis of Readout Strategies
The selection of a readout strategy is critical and depends on the required sensitivity, speed, cost, and the need for quantitative results. The table below summarizes the key characteristics of the four methods discussed.
Table 1: Comparative Analysis of Biosensor Signal Readout Strategies for Microbial Detection
| Readout Strategy | Typical Detection Mechanism | Approx. Detection Time | Key Advantages | Inherent Limitations | Example Application in Fermentation |
|---|---|---|---|---|---|
| Fluorescence | Emission of light upon excitation of a fluorophore [49] | Minutes to a few hours [50] | Very high sensitivity, multiplexing capability, quantitative | Requires excitation source/ detector, can be affected by environmental quenching | In-situ detection of specific probiotic strains (e.g., Lactobacillus spp.) in yogurt fermentation [18] |
| Colorimetric | Visual color change measurable by absorbance/reflectance [49] | Minutes to hours [49] | Simple, low-cost, instrument-free (visually read), semi-quantitative with scanners | Lower sensitivity than fluorescence/ electrochemical, subject to subjective visual interpretation | Monitoring spoilage organisms (e.g., Pseudomonas) in meat via volatile amine sensors [18] |
| Lateral Flow (LF) | Capillary flow with capture lines, typically colorimetric or electrochemical [51] | < 30 minutes [51] | Rapid, user-friendly, portable, low cost, high stability | Generally semi-quantitative, lower sensitivity than lab-based methods | Point-of-care detection of Salmonella contamination in fermented sausage production environments [18] [51] |
| Electrochemical (EC) | Measurement of electrical changes (current, potential, impedance) [18] [50] | 20 minutes - 4 hours [18] [50] | High sensitivity, excellent for quantification, miniaturization, compatible with complex matrices | Signal drift, electrode fouling, often requires sophisticated data interpretation | Real-time tracking of L. plantarum activity and pH changes during fermentation [18] [50] |
Detailed Experimental Protocols
Electrochemical CRISPR/Cas (EC-CRISPR) Biosensor for Pathogen Detection
This protocol outlines the steps for detecting a specific bacterial pathogen (e.g., Salmonella Typhimurium) in a fermentation sample using an EC-CRISPR/Cas12a biosensor, achieving high sensitivity through signal amplification [50].
Workflow Overview:
Materials:
- CRISPR/Cas12a protein: The core effector protein for specific DNA target recognition and subsequent reporter cleavage [50].
- crRNA: Custom-designed guide RNA complementary to the target pathogen's DNA sequence (e.g., a region of the S. Typhimurium genome) [50].
- Saltatory Rolling Circle Amplification (SRCA) reagents: For isothermal pre-amplification of the target nucleic acid to enhance sensitivity [50].
- ssDNA-Fc reporter probe: A single-stranded DNA molecule labeled with a Ferrocene (Fc) redox tag. The Cas12a protein cleaves this reporter upon target activation [50].
- Screen-printed gold or carbon electrode: The transducer surface for immobilizing the reporter probe and measuring the electrochemical signal [50].
- Potentiostat: The instrument for applying potential and measuring the resulting current (e.g., via Square Wave Voltammetry) [50].
Procedure:
- Sample Preparation: Extract total nucleic acids from the fermented food matrix (e.g., kimchi, sausage) using a commercial kit.
- Target Amplification: Perform SRCA amplification using specific primers for the target pathogen. This step enriches the target sequence, enabling ultra-sensitive detection.
- CRISPR/Cas12a Reaction:
- Prepare the reaction mix containing Cas12a protein, crRNA, and the ssDNA-Fc reporter probe.
- Add the amplified SRCA product to the reaction mix.
- Incubate at 37°C for 15-30 minutes. If the target DNA is present, Cas12a becomes activated and cleaves the ssDNA-Fc reporter.
- Electrode Measurement:
- Immobilize the cleavage reaction mixture onto the working electrode surface.
- Using a potentiostat, perform Square Wave Voltammetry (SWV) to measure the redox current from the intact Fc tag. Extensive reporter cleavage leads to a decrease in the measured current.
- Data Analysis: Quantify the target concentration by comparing the signal suppression to a standard curve generated with known concentrations of the target DNA.
Fluorescence-Based Wearable Sensor for Metabolite Monitoring
This protocol describes a method for continuous, non-invasive monitoring of microbial metabolites in sweat using a flexible, microfluidic fluorescence sensor, which can be adapted for workers in fermentation facilities to monitor environmental microbial activity [49].
Workflow Overview:
Materials:
- Polydimethylsiloxane (PDMS) substrate: A flexible, biocompatible polymer used to create the microfluidic channels and chambers [49].
- Fluorescent probes: Molecules that selectively bind to target analytes (e.g., chloride, sodium, zinc) and undergo a change in fluorescence intensity [49].
- Miniaturized LED source: Integrated into the wearable device to excite the fluorescent probes at a specific wavelength [49].
- Smartphone with optical module: Acts as a portable detector to capture the emitted fluorescence light and an analyzer to quantify the intensity [49].
Procedure:
- Sensor Fabrication: Fabricate a microfluidic device from PDMS using soft lithography. The device should contain networks of microchannels and reservoirs pre-loaded with the specific fluorescent probes for the target metabolites.
- Sweat Sampling: Affix the wearable sensor to the skin. As sweat is naturally produced, it is drawn into the microfluidic channels via capillary action and comes into contact with the pre-loaded probes.
- Analyte-Probe Reaction: The target analytes in the sweat (e.g., chloride ions) bind to their respective fluorescent probes, causing a quantifiable change (e.g., quenching) in the probe's fluorescence.
- Optical Measurement: The integrated LED excites the probes. The smartphone camera, housed in a custom-made optical module that blocks ambient light, captures the resulting fluorescence emission.
- Data Quantification: A dedicated smartphone application processes the captured image, correlating the fluorescence intensity to the analyte concentration using a pre-calibrated curve.
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Reagents and Materials for Biosensor Development
| Item/Category | Function/Description | Specific Application Example |
|---|---|---|
| CRISPR Effector Proteins (Cas12a, Cas13a) | Recognize specific nucleic acid sequences and exhibit trans-cleavage activity upon activation, providing the basis for signal amplification [50]. | Cas12a for DNA targets (bacterial contamination), Cas13a for RNA targets (viral contamination or specific microbial gene expression) [50]. |
| Aptamers | Synthetic single-stranded DNA or RNA oligonucleotides that bind to specific non-nucleic acid targets (proteins, toxins) with high affinity [50]. | Detecting microbial toxins (e.g., Staphylococcal enterotoxins) in fermented products without needing nucleic acid extraction [50]. |
| Screen-Printed Electrodes (SPE) | Disposable, low-cost electrodes that serve as the solid support for biorecognition events and electrochemical signal transduction [50] [51]. | Customizable platforms for electrochemical CRISPR biosensors; can be integrated into lateral flow strips for electrochemical Lateral Flow Assays (eLFAs) [51]. |
| Redox Reporters (e.g., Ferrocene, Methylene Blue) | Molecules that undergo reversible oxidation/reduction reactions, producing a measurable electrochemical current [50]. | Tagging ssDNA reporters in EC-CRISPR assays. Cleavage of the reporter changes the electron transfer efficiency, modulating the signal [50]. |
| Flexible Polymer Substrates (e.g., PDMS, PET) | Provide a flexible, stretchable, and biocompatible base for constructing wearable and point-of-care biosensors [49]. | Used in wearable microfluidic sweat sensors for metabolite monitoring and in smart contact lenses for tear fluid analysis [49]. |
The convergence of CRISPR biology with advanced signal readout technologies is forging a new paradigm in food fermentation research. The choice of readout strategy—whether for the ultimate sensitivity of electrochemical detection, the visual simplicity of colorimetric and lateral flow assays, or the precise quantification of fluorescence—directly empowers researchers to monitor and control microbial processes with unprecedented precision. As these technologies continue to evolve, particularly with the integration of artificial intelligence for data analysis and the development of wearable form factors, they promise to usher in an era of fully intelligent, automated, and sustainable fermentation processes, ensuring the production of safe, high-quality fermented foods.
Within food fermentation research, ensuring microbial purity is paramount. The sensitive and specific detection of foodborne pathogens such as Listeria monocytogenes, Salmonella spp., and Escherichia coli O157:H7 in complex matrices like fermenting substrates presents a significant analytical challenge. Traditional culture methods are time-consuming, while conventional molecular techniques often lack the required sensitivity or portability for in-process monitoring. CRISPR (Clustered Regularly Interspaced Short Palindromic Repeats)-based biosensors have emerged as a revolutionary tool, offering rapid, highly sensitive, and specific detection of pathogen nucleic acids. This application note details protocols and data for utilizing these biosensors, with a particular focus on their integration into food fermentation safety assurance programs.
Performance of CRISPR-Based Biosensors for Pathogen Detection
The following table summarizes the experimental performance of recently developed CRISPR/Cas systems for detecting key foodborne pathogens, demonstrating their applicability in complex food samples.
Table 1: Analytical Performance of CRISPR-Based Biosensors for Foodborne Pathogen Detection
| Target Pathogen | CRISPR System & Amplification | Sample Matrix | Limit of Detection (LOD) | Time-to-Result | Specificity (Cross-reactivity) | Citation |
|---|---|---|---|---|---|---|
| Listeria monocytogenes | Cas12a / LAMP | Enoki mushroom | 100 CFU/g | <2 hours | No cross-reactivity with 5 other Listeria spp. and 14 other common pathogenic bacteria | [52] |
| Listeria monocytogenes | Cas12a / RAA | Pure culture & beef | 350 CFU/mL (Pure culture); 2.3 CFU/25g (after enrichment in beef) | ~2 hours (incl. enrichment) | Specific for 13 L. monocytogenes strains; no cross-reactivity with 5 other Listeria spp. and 14 non-Listeria pathogens | [53] |
| Salmonella typhimurium | dCas9 / LAMP (SCOUT-dCas9) | Contaminated real food samples | 1 CFU/mL | Information not specified in abstract | Satisfactory selectivity | [54] |
| Salmonella spp. | Cas13a / RPA | Clinical stool samples | 100 copies (Two-step method) | 45 min (Two-step); 20 min (One-tube) | Highly specific; no cross-reaction with nine other diarrheal bacteria | [55] |
Detailed Experimental Protocols
Protocol A: Detection ofListeria monocytogenesusing LAMP-CRISPR/Cas12a with Magnetic Pre-concentration
This protocol leverages tetraethylenepentamine (TEPA)-functionalized magnetic nanoparticles (MNPs) for pre-concentration to enhance sensitivity in complex food matrices, followed by LAMP amplification and Cas12a-mediated detection [52].
I. Materials and Reagents
- TEPA-functionalized MNPs: For electrostatic capture of negatively charged bacterial cells.
- LAMP Primer Mix: Specific for the hly gene of L. monocytogenes.
- Cas12a Nuclease: Effector protein for target-specific cis-cleavage and non-specific trans-cleavage of ssDNA reporters.
- crRNA: Designed to target a specific region within the hly gene amplicon.
- ssDNA Fluorescent Reporter: e.g., FAM-TTATT-BHQ1 quenched fluorescent probe.
- LAMP Reagents: Including Bst DNA polymerase, dNTPs, and isothermal amplification buffer.
- Food Sample: Homogenized in an appropriate buffer (e.g., 20 mL).
II. Step-by-Step Procedure
- Pathogen Capture and Concentration:
- Add TEPA-functionalized MNPs to 20 mL of homogenized food sample.
- Incubate with constant mixing for a predetermined optimal time (e.g., 30-60 minutes) to allow bacteria to bind to MNPs.
- Apply a magnetic field to concentrate the MNP-bacteria complexes. Carefully discard the supernatant.
- Resuspend the magnetic pellet in a small volume (e.g., 100 μL) of elution buffer for lysis and DNA release.
Nucleic Acid Amplification via LAMP:
- Use the extracted DNA or crude lysate as a template in the LAMP reaction.
- Prepare the LAMP reaction mixture containing primer mix, Bst polymerase, dNTPs, and buffer.
- Incubate the reaction at a constant temperature of 60-65°C for 45-60 minutes.
- Terminate the reaction by heating at 80°C for 5 minutes.
CRISPR/Cas12a-mediated Detection:
- Prepare the Cas12a detection cocktail containing Cas12a protein, hly-specific crRNA, and the fluorescent ssDNA reporter.
- Mix the LAMP amplification product with the detection cocktail.
- Incubate at 37-42°C for 10-20 minutes to allow for Cas12a activation and reporter cleavage.
- Visualize the fluorescence signal using a portable fluorometer or a blue light transilluminator. A positive result is indicated by a significant increase in fluorescence.
Diagram 1: LAMP-CRISPR/Cas12a detection workflow with magnetic pre-concentration.
Protocol B: One-Tube RPA-CRISPR/Cas13a forSalmonellaspp. Detection
This protocol describes a rapid, one-tube assay for Salmonella detection targeting the invA gene, combining Recombinase Polymerase Amplification (RPA) and Cas13a's collateral RNAse activity, suitable for point-of-care testing [55].
I. Materials and Reagents
- RPA Reagents: TwistAmp basic rehydration buffer, magnesium acetate, forward and reverse primers specific to the invA gene.
- Cas13a Protein: Purified Leptotrichia wadei Cas13a (LwCas13a).
- crRNA: Specific for the invA gene amplicon.
- RNA Reporter Probe: Fluorescently quenched RNA oligonucleotide (e.g., FAM-labeled).
- NTP Mix: ATP, GTP, CTP, UTP.
II. Step-by-Step Procedure
- Reaction Setup:
- Prepare a master mix containing RPA rehydration buffer, RPA primers, Cas13a protein, invA-specific crRNA, the RNA reporter probe, and NTPs.
- Add the template DNA (extracted from the sample) to the master mix.
- Transfer the mixture to a single reaction tube.
- Initiate the reaction by adding magnesium acetate.
Isothermal Amplification and Detection:
- Incubate the reaction tube at 39°C for 20 minutes in a fluorescence reader or a heated block.
- During this single step, two reactions occur concurrently:
- RPA: Isothermal amplification of the invA gene target.
- CRISPR/Cas13a Activation: The RPA amplicon is recognized by the Cas13a/crRNA complex, activating its trans-cleavage activity, which cleaves the RNA reporter and generates a fluorescent signal.
Result Interpretation:
- Monitor the fluorescence in real-time. A rapid increase in fluorescence within the 20-minute incubation indicates a positive result for Salmonella spp.
Diagram 2: One-tube RPA-CRISPR/Cas13a detection workflow for Salmonella.
The Scientist's Toolkit: Essential Research Reagents
Table 2: Key Reagent Solutions for CRISPR-Based Pathogen Detection
| Reagent / Material | Function / Role in the Assay | Examples / Specifications |
|---|---|---|
| Cas Effector Proteins | The core nuclease that, upon target recognition, is activated for specific cleavage and/or collateral trans-cleavage of reporters. | Cas12a (for DNA targets), Cas13a (for RNA targets), dCas9 (for binding without cleavage) [56] [54]. |
| crRNA / sgRNA | Guides the Cas protein to the specific target nucleic acid sequence with high precision, ensuring assay specificity. | A 20-30 nt spacer sequence complementary to the target gene (e.g., hly, invA) [53] [55]. |
| Isothermal Amplification Kits | Rapidly amplifies the target nucleic acid at a constant temperature, eliminating the need for a thermal cycler. | LAMP (Loop-mediated Isothermal Amplification), RPA (Recombinase Polymerase Amplification), RAA (Recombinase Aided Amplification) [53] [52] [55]. |
| Nucleic Acid Reporters | Generates a detectable signal (fluorescent, colorimetric) upon trans-cleavage by an activated Cas protein. | ssDNA probes for Cas12a (e.g., FAM-TTATT-BHQ1); RNA probes for Cas13a [53] [52] [55]. |
| Functionalized Magnetic Nanoparticles (MNPs) | Captures and concentrates target pathogens from large-volume food samples, improving sensitivity and reducing interference. | TEPA-functionalized MNPs (electrostatic binding), antibody-conjugated MNPs (immunomagnetic separation) [52]. |
| Portable Detection Devices | Enables visual or instrument-based signal readout at the point-of-need (e.g., in a production facility). | Portable fluorometers, blue light transilluminators, lateral flow dipsticks [57] [58]. |
Monitoring and Quantifying Starter Cultures (e.g., Lactobacillus, Streptococcus) for Process Control
Within food fermentation research, the precise monitoring and quantification of starter cultures, such as Lactobacillus and Streptococcus, is critical for ensuring consistent product quality, safety, and process efficiency. Traditional culture-based methods and even modern PCR techniques are often limited by time, required expertise, or complex instrumentation [18] [5]. CRISPR-based biosensors represent a transformative toolset, offering rapid, sensitive, and specific detection that is ideally suited for real-time process control in industrial fermentation. These systems leverage the programmable nature of CRISPR-Cas proteins to target specific genetic signatures of starter cultures, enabling precise quantification directly within complex food matrices [59] [60]. This application note details the integration of these biosensors into fermentation research and development pipelines.
Core Mechanism of CRISPR-Cas Biosensors for Nucleic Acid Detection
CRISPR-based diagnostics utilize Cas effector proteins that are guided by a custom-designed CRISPR RNA (crRNA) to find and bind to specific nucleic acid target sequences. Upon target recognition, certain Cas proteins exhibit collateral activity, non-specifically cleaving surrounding reporter molecules to generate a detectable signal [34] [5]. This combination of precise targeting and amplified signal output is the foundation of their utility in starter culture monitoring.
Table 1: Key CRISPR-Cas Systems for Biosensing in Fermentation
| Cas Protein | Target Type | PAM/PFS Requirement | Collateral Activity | Ideal Starter Culture Application |
|---|---|---|---|---|
| Cas9 | dsDNA | 5'-NGG | None | Target enrichment; multiplexed detection via dCas9 [60] |
| Cas12 (e.g., Cas12a) | dsDNA, ssDNA | 5'-TTTV | ssDNA cleavage | Quantifying bacterial load (DNA targets) [60] [5] |
| Cas13 (e.g., Cas13a) | RNA | 3'-H (A, U, C) | ssRNA cleavage | Monitoring metabolic activity (rRNA/mRNA targets) [60] [34] |
The following diagram illustrates the fundamental signaling pathways for the two primary Cas systems used in quantification.
Experimental Protocol for QuantifyingLactobacillusspp. Using CRISPR-Cas12a
This protocol provides a step-by-step methodology for the absolute quantification of Lactobacillus species in a fermented milk sample using a Cas12a-based biosensor coupled with recombinase polymerase amplification (RPA).
Sample Preparation and Nucleic Acid Extraction
- Sample Collection: Aseptically collect 1 mL of fermenting milk product at the desired time point.
- Cell Lysis: Mix the sample with 2 mL of lysis buffer (e.g., containing lysozyme and proteinase K) and incubate at 55°C for 30 minutes.
- DNA Extraction: Purify genomic DNA using a commercial silica-column or magnetic bead-based kit. Elute the DNA in 50-100 µL of nuclease-free water.
- DNA Quantification: Measure the concentration and purity (A260/A280 ratio) of the extracted DNA using a spectrophotometer. Note: This step is for quality control and does not provide species-specific quantification.
Isothermal Amplification (RPA)
- Prepare RPA Reaction Mix (50 µL total volume):
- Recombinase Polymerase Amplification (RPA) dry pellet
- 2.4 µL of each primer (10 µM) targeting the Lactobacillus 16S rRNA gene.
- 1 µL of extracted DNA template.
- Nuclease-free water to 50 µL.
- Amplification: Incubate the reaction at 39°C for 15-20 minutes. No thermal cycler is required.
CRISPR-Cas12a Detection
- Prepare Cas12a Detection Mix (20 µL total volume):
- 100 nM purified Cas12a enzyme.
- 120 nM crRNA designed to target the internal sequence of the RPA amplicon.
- 200 nM ssDNA fluorescent reporter (e.g., FAM-TTATT-BHQ1).
- 1x Cas12a reaction buffer.
- Initiate Reaction: Add 5 µL of the RPA amplicon product directly into the Cas12a detection mix.
- Incubate and Detect: Incubate the combined reaction at 37°C for 10-30 minutes. Monitor fluorescence in real-time using a portable fluorometer or plate reader (Excitation: 485 nm, Emission: 528 nm).
Data Analysis and Quantification
- Generate Standard Curve: Using the same protocol, run reactions with a dilution series of genomic DNA from a Lactobacillus strain with a known concentration (e.g., CFU/mL).
- Plot Results: Plot the log of the initial DNA concentration (or CFU) against the time-to-positive (TTP) or the fluorescence intensity at a fixed endpoint.
- Interpolate Unknowns: Use the standard curve equation to calculate the concentration of Lactobacillus in the test samples based on their fluorescence signal.
Performance Data and Validation
Validation against traditional culture methods is essential for establishing the reliability of CRISPR-based biosensors.
Table 2: Performance Metrics of a CRISPR-Cas12a Biosensor for S. thermophilus
| Parameter | Result | Comparative Method (Plate Count) | Notes |
|---|---|---|---|
| Detection Limit | 10² CFU/mL [5] | 10¹-10² CFU/mL (after enrichment) | Sufficient for monitoring active fermentation. |
| Total Assay Time | 45-60 minutes [5] | 24-48 hours | Enables near real-time process decisions. |
| Dynamic Range | 10² - 10⁸ CFU/mL | 10¹ - 10⁹ CFU/mL | Covers typical working ranges for starter cultures. |
| Specificity | 100% for target species [60] | High, but can be laborious | No cross-reactivity with non-target lactic acid bacteria. |
| Signal-to-Noise Ratio | >20:1 | Not Applicable | Clear distinction between positive and negative signals. |
The Scientist's Toolkit: Key Research Reagent Solutions
Table 3: Essential Reagents for CRISPR-Based Starter Culture Monitoring
| Reagent / Material | Function / Description | Example / Specification |
|---|---|---|
| crRNA | Guides Cas protein to the specific target sequence; defines specificity. | Synthetic RNA oligo designed against conserved region of 16S rRNA or species-specific gene. |
| Cas Effector Protein | The enzyme that executes target binding and collateral cleavage. | Purified Cas12a (Cpf1) or Cas13a; commercial lyophilized formats available for stability. |
| Isothermal Amplification Kit | Rapidly amplifies the target nucleic acid sequence at a constant temperature. | Recombinase Polymerase Amplification (RPA) or LAMP kits, including primers. |
| Fluorescent Reporter | A cleavable molecule that emits fluorescence upon collateral activity, providing the signal. | ssDNA reporter (for Cas12) or ssRNA reporter (for Cas13) with a fluorophore-quencher pair. |
| Nucleic Acid Extraction Kit | Isolates high-purity DNA/RNA from complex fermentation matrices. | Kits optimized for Gram-positive bacteria (e.g., with enhanced lysozyme digestion steps). |
| Portable Fluorometer | Enables on-site, real-time signal detection for process control. | Battery-operated device with appropriate filters for the fluorophore used (e.g., FAM). |
Integrated Workflow for Fermentation Process Control
Implementing this technology effectively requires a seamless workflow from sampling to data interpretation, as summarized below.
Implementation Considerations
- crRNA Design: Select target sequences that are unique to the starter culture of interest to avoid cross-reactivity with background microbiota. The 16S rRNA gene is a common target, but functional genes can provide higher specificity [34].
- Matrix Effects: Complex food matrices (e.g., high fat or protein content) can inhibit enzymatic reactions. Optimization of sample dilution or inclusion of amplification enhancers (e.g., BSA) is often necessary [5].
- Quantification Approach: For absolute quantification, a standard curve is required. For relative quantification or presence/absence testing, the protocol can be simplified.
- Multiplexing Potential: Using multiple crRNAs or different Cas proteins (e.g., Cas12 and Cas13 simultaneously) allows for the parallel monitoring of different bacterial strains in a co-culture fermentation [59] [5].
Within food fermentation research, the precise tracking of spoilage microorganisms and their undesirable metabolic outputs is crucial for ensuring product quality and safety. CRISPR-based biosensors have emerged as powerful tools for this purpose, offering researchers unparalleled specificity and sensitivity in detecting both microbial nucleic acids and associated virulence factors. This application note details the implementation of CRISPR-Cas biosensing platforms for monitoring spoilage targets, providing structured experimental protocols and reagent solutions to accelerate adoption in food microbiology research.
Core Detection Mechanisms and Targets
CRISPR-based detection leverages the programmable nature of Cas proteins to identify specific nucleic acid sequences with single-base pair resolution. The table below summarizes the primary CRISPR systems employed in spoilage microbe tracking and their characteristic targets.
Table 1: CRISPR Systems for Tracking Spoilage Microbes and Metabolic Activity
| CRISPR System | Target Type | Cleavage Activity | Primary Spoilage Targets | Detection Examples |
|---|---|---|---|---|
| Cas9 (Type II) | dsDNA | cis-cleavage only | Species-specific genes for spoilage bacteria identification | Listeria spp., Pseudomonas spp. [60] [6] |
| Cas12 (Type V) | dsDNA, ssDNA | cis- and trans-cleavage | Virulence genes, antibiotic resistance markers, species-specific DNA | Salmonella virulence factors, E. coli O157:H7 [32] [60] [6] |
| Cas13 (Type VI) | RNA | cis- and trans-cleavage | Metabolic gene transcripts (e.g., toxin production), bacterial mRNA | Quorum sensing signaling molecules, spoilage metabolite genes [32] [60] |
The trans-cleavage activity of Cas12 and Cas13 proteins is particularly valuable for biosensing. Upon recognition of its specific target, the Cas protein becomes activated to indiscriminately cleave nearby reporter molecules, enabling significant signal amplification and highly sensitive detection [32] [60] [6].
Experimental Protocol: Cas12a-based Detection of Spoilage Bacteria
This protocol provides a detailed methodology for detecting specific spoilage bacteria (e.g., Pseudomonas spp.) in a fermented meat model system using a Cas12a-based fluorescence biosensor.
The following diagram illustrates the complete experimental workflow from sample preparation to result interpretation:
Materials and Equipment
- Biological Samples: Artificially inoculated meat samples (e.g., 25 g ground meat with 10⁸ CFU/g Pseudomonas)
- DNA Extraction Kit: Commercial kit (e.g., QIAGEN DNeasy Blood & Tissue Kit)
- Amplification Reagents: LAMP or RPA kit with specific primers targeting the Pseudomonas 16S rRNA gene
- CRISPR Reaction Components:
- Recombinant LbaCas12a enzyme (100 nM)
- Custom crRNA (50 nM) targeting Pseudomonas 16S rRNA sequence
- ssDNA-FQ reporter (500 nM, 6-FAM/TAMRA-labeled)
- NEBuffer 2.1 (1X)
- Equipment: Real-time PCR thermocycler or fluorescence plate reader, microcentrifuge, vortex mixer, nanodrop spectrophotometer
Step-by-Step Procedure
Sample Preparation and DNA Extraction
- Homogenize 25 g of meat sample in 225 mL of sterile buffered peptone water using a stomacher for 2 minutes.
- Centrifuge homogenate at 12,000 × g for 5 minutes to pellet bacterial cells.
- Extract genomic DNA from the pellet using a commercial kit according to the manufacturer's instructions.
- Quantify DNA concentration and purity using a nanodrop spectrophotometer (A260/A280 ratio of ~1.8 indicates pure DNA).
Isothermal Amplification (Using LAMP)
- Prepare 25 µL LAMP reaction mixture containing:
- 1X Isothermal Amplification Buffer
- 6 mM MgSO₄
- 1.4 mM each dNTP
- 0.2 µM each F3/B3 primer
- 1.6 µM each FIP/BIP primer
- 8 U Bst 2.0 WarmStart DNA Polymerase
- 5 µL template DNA
- Incubate reaction at 65°C for 30-60 minutes, then heat-inactivate at 80°C for 5 minutes.
- Prepare 25 µL LAMP reaction mixture containing:
CRISPR-Cas12a Detection
- Prepare 20 µL CRISPR reaction mix containing:
- 1X NEBuffer 2.1
- 100 nM LbaCas12a
- 50 nM Pseudomonas-specific crRNA
- 500 nM ssDNA-FQ reporter
- 2 µL of the LAMP amplification product
- Incubate the reaction at 37°C for 30 minutes.
- Prepare 20 µL CRISPR reaction mix containing:
Signal Measurement and Data Analysis
- Measure fluorescence intensity (excitation/emission: 485/535 nm) using a plate reader at 5-minute intervals during the 37°C incubation.
- Calculate the fluorescence fold change relative to the negative control (non-inoculated sample).
- A sample is considered positive if the fluorescence signal exceeds the threshold (mean + 3 standard deviations of the negative control) within 30 minutes.
Critical Steps and Troubleshooting
- crRNA Design: Ensure the crRNA spacer sequence is complementary to the target region and the Cas12a PAM sequence (5'-TTTV-3') is present adjacent to the target site [60].
- Amplification Contamination: Use separate work areas for pre- and post-amplification steps and include negative controls to monitor for contamination.
- Signal Optimization: If fluorescence signal is weak, titrate the crRNA concentration (20-100 nM) and extend the Cas12a reaction incubation time.
The Scientist's Toolkit: Research Reagent Solutions
The table below outlines essential reagents and their functions for establishing CRISPR-based detection of spoilage microbes.
Table 2: Key Research Reagent Solutions for CRISPR-Based Spoilage Tracking
| Reagent / Material | Function | Example Specifications |
|---|---|---|
| Cas Proteins | Target recognition and nucleic acid cleavage | Recombinant Cas12a (LbaCas12a, 100 nM), Cas13a (LwaCas13a, 50 nM) [60] [6] |
| Guide RNAs | Sequence-specific targeting | crRNA for Cas12a (50 nM); crRNA for Cas13a (50 nM), HPLC-purified [60] [6] |
| Reporter Probes | Signal generation upon trans-cleavage | ssDNA-FQ reporter for Cas12 (500 nM); ssRNA-FQ reporter for Cas13 (500 nM) [32] [60] |
| Isothermal Amplification Kits | Target pre-amplification | LAMP or RPA kits for signal enhancement [60] [6] |
| Fluorescence Detector | Signal quantification | Real-time PCR instrument or portable fluorescence reader [61] [6] |
Detection Mechanism and Signal Transduction
The core mechanism of Cas12a-based detection involves target-binding-induced collateral cleavage of a reporter molecule, as illustrated below:
Data Interpretation and Analysis
CRISPR-based detection enables both qualitative identification and quantitative assessment of spoilage microbes. The typical output is a fluorescence growth curve where the time to reach a threshold fluorescence (Time-to-Positive, TTP) is inversely correlated with the initial target concentration. Standard curves generated using samples with known bacterial concentrations (e.g., 10¹ to 10⁸ CFU/mL) allow for quantification of unknown samples. This system can achieve high sensitivity, detecting targets at attomolar (aM) concentrations with single-base specificity [22] [60] [6].
Navigating Practical Hurdles: Optimization for Real-World Fermentation Environments
The analysis of microbial contaminants in fermented foods using CRISPR-based biosensors presents a significant challenge due to matrix interference. Complex food components, including fats, proteins, and carbohydrates, can obstruct target recognition, quench signal output, and ultimately compromise detection accuracy and sensitivity [62] [63]. This application note details the specific mechanisms of matrix interference and provides validated protocols to mitigate these effects, ensuring reliable pathogen detection in complex fermentation matrices.
The food matrix refers to the intricate physical and chemical structure of food, where nutrients and other components are organized and interact in ways that influence how they behave during analysis [64]. In fermented products, this matrix is particularly complex, often containing a diverse microbial consortium, variable pH, and a rich blend of organic acids and enzymes, all of which can interfere with biosensor function [63].
Mechanisms of Matrix Interference
Understanding how different food components interfere with biosensing is crucial for developing effective countermeasures. The table below summarizes the primary interference mechanisms.
Table 1: Mechanisms of Matrix Interference in Food Biosensing
| Matrix Component | Primary Sources in Fermented Foods | Interference Mechanisms | Impact on CRISPR Biosensors |
|---|---|---|---|
| Fats & Lipids | Dairy (cheese, yogurt), fermented meats | • Non-specific adsorption to sensors [63]• Formation of viscous microenvironments [65]• Alters protein structure & accessibility [65] | Reduced collision efficiency, quenched signal output, altered Cas enzyme activity |
| Proteins | Dairy, soy products, meat ferments | • Binds non-specifically to guide RNA or Cas proteins [62]• Competes for binding sites on sensor surfaces [63]• Forms aggregates that scatter light [65] | False positives/negatives, reduced signal-to-noise ratio, physical blocking of reaction sites |
| Carbohydrates | Fermented vegetables, sourdough, beers | • Increases sample viscosity, slowing diffusion [62] |
Slows reaction kinetics, inhibits enzyme co-factors, quenches fluorescent signals |
| Complex Matrices | All fermented foods | • Combined effects of above components [62]• Endogenous enzymes (e.g., proteases, nucleases) degrade biosensor components [63] | Synergistic degradation of assay components, leading to significant signal loss |
Strategies for Mitigating Matrix Effects
Sensor Design with Built-In Resilience
A promising approach to circumvent matrix interference involves the strategic design of the biosensing system itself. The CRISPR/Cas12a-mediated enzymatic cascade reaction Magnetic Relaxation Switching (CMCR-MRS) biosensor exemplifies this strategy [66] [67]. This system minimizes background interference by employing a paramagnetic ion-mediated signal readout, which depends on the valence state conversion of Mn(VII) to Mn(II), rather than on changes in physical aggregation of nanoparticles that can be non-specifically influenced by matrix components [66]. This design offers superior biocompatibility and minimizes background interference in complex samples [66].
Sample Processing and Pre-Treatment
Effective sample preparation is critical to isolate the target analyte from interfering substances.
- Dilution: Simple dilution of the sample can reduce the concentration of interferents below a problematic threshold, though this may also dilute the target pathogen.
- Filtration: Centrifugal filters can remove large proteins and particulate matter.
- Chemical Pre-Treatment: Use of solvents to precipitate proteins or detergents to solubilize lipids can clean up samples prior to analysis.
Protocols for Overcoming Interference in Fermented Food Analysis
Protocol: CMCR-MRS for Detection in High-Fat Matrices
This protocol is adapted from the CMCR-MRS biosensor developed for Salmonella typhimurium and is effective for high-fat matrices like cheese or fermented sausage [66] [67].
Principle: CRISPR/Cas12a targets pathogen DNA, triggering trans-cleavage activity that releases Alkaline Phosphatase (ALP) from magnetic nanoparticles. The free ALP catalyzes the generation of ascorbic acid, which reduces paramagnetic Mn(VII) to Mn(II), causing a measurable change in the transverse relaxation time (T2) [66].
Workflow: The following diagram illustrates the CMCR-MRS protocol for detecting pathogens in high-fat food matrices.
Materials & Reagents:
- Lysis Buffer: For DNA release via thermal lysis.
- MNP-ALP Probes: Magnetic nanoparticles conjugated to ALP via ssDNA linkers.
- CRISPR/Cas12a-crRNA RNP Complex: Pre-complexed Cas12a with crRNA targeting the pathogen of interest.
- AAP Solution: 2-Phospho-L-ascorbic acid trisodium salt (substrate for ALP).
- KMnO₄ Solution: Source of paramagnetic Mn(VII) ions.
- Low-Field NMR Spectrometer: For measuring transverse relaxation time (T2).
Procedure:
- Sample Homogenization: Homogenize 1 g of food sample in 10 mL of PBS buffer.
- Target DNA Release: Centrifuge the homogenate and subject the pellet to thermal lysis (95°C for 10 minutes) to release genomic DNA.
- CRISPR Cleavage Reaction:
- In a reaction tube, combine:
- 10 µL of the extracted DNA supernatant.
- 5 µL of Cas12a-crRNA RNP complex.
- 10 µL of MNP-ALP probes.
- 25 µL of reaction buffer.
- Incubate at 37°C for 30 minutes.
- In a reaction tube, combine:
- Enzymatic Cascade:
- Add 20 µL of AAP solution to the reaction mixture.
- Incubate at 37°C for 20 minutes. The released ALP will dephosphorylate AAP to generate ascorbic acid.
- Magnetic Relaxation Switching:
- Add 30 µL of KMnO₄ solution to the mixture.
- Incubate for 5 minutes. Ascorbic acid reduces Mn(VII) to Mn(II).
- Detection:
- Transfer the solution to an NMR tube.
- Measure the transverse relaxation time (T2) using the low-field NMR spectrometer.
- The change in T2 (ΔT2) is proportional to the target pathogen concentration.
The Scientist's Toolkit: Key Research Reagents
Table 2: Essential Reagents for CRISPR-Based Detection in Complex Matrices
| Reagent / Material | Function | Consideration for Matrix Interference |
|---|---|---|
| Cas12a Enzyme | Target-specific cleavage and trans-nuclease activity. | High-purity grades minimize non-specific activation by matrix proteases. |
| crRNA | Guides Cas enzyme to the target DNA sequence. | Meticulous design is required to avoid off-target binding to non-pathogen DNA in the matrix. |
| Magnetic Nanoparticles | Solid support for probes; enables separation and concentration. | Surface coating (e.g., PEG) can reduce non-specific binding from fats and proteins [66]. |
| Paramagnetic Ions (Mn(VII)/Mn(II)) | Core of the MRS signal switch. | Less susceptible to aggregation-based false signals compared to nanoparticle aggregation methods [66]. |
| Alkaline Phosphatase (ALP) | Enzyme for signal amplification cascade. | Check for endogenous ALP activity in the food matrix that may cause high background. |
Matrix interference from fats, proteins, and carbohydrates represents a formidable barrier to the accurate detection of pathogens in fermented foods. The CMCR-MRS biosensor demonstrates that a combination of robust sensor design, which uses paramagnetic ion conversion for readout, and optimized sample handling protocols can effectively overcome these challenges. This approach provides a reliable pathway for employing sensitive, amplification-free CRISPR-based diagnostics in complex fermentation research, ensuring that results truly reflect the microbial safety of the product.
Strategies for Streamlined Sample Preparation and Nucleic Acid Extraction
In the field of food fermentation research, the accurate and timely detection of microbial contaminants is crucial for ensuring product quality and safety. CRISPR-based biosensors have emerged as powerful tools for molecular diagnostics, offering unparalleled specificity and sensitivity in identifying pathogenic microorganisms [68] [34]. However, the performance of these advanced detection systems is fundamentally dependent on the efficiency of upstream sample preparation and nucleic acid extraction processes. Effective sample preparation must address complex food matrices, inhibit PCR inhibitors, and yield high-quality nucleic acids suitable for downstream CRISPR-based detection [50] [69]. This application note provides detailed protocols and strategies for streamlined sample preparation and nucleic acid extraction, specifically optimized for integration with CRISPR-based biosensing platforms in food fermentation research.
Core Principles of CRISPR-Based Detection
Molecular Mechanisms of CRISPR Systems
CRISPR-Cas systems function through RNA-guided recognition and cleavage of nucleic acid targets. The system utilizes Cas proteins that become activated upon binding to target sequences complementary to their guide RNAs [68] [34]. Key CRISPR systems used in biosensing include:
- Cas12a: Targets DNA sequences and exhibits trans-cleavage activity against single-stranded DNA (ssDNA) upon activation [19]. It recognizes T-rich PAM sequences and requires only a single crRNA for operation [60].
- Cas13a: Targets RNA sequences and demonstrates trans-cleavage activity against single-stranded RNA (ssRNA) [68] [70]. This system is particularly valuable for detecting RNA viruses or monitoring gene expression in fermentation processes.
- Cas9: Primarily used for gene editing applications but can be adapted for diagnostic purposes through engineered versions such as dead Cas9 (dCas9) which retains target binding ability without cleavage activity [70].
The collateral trans-cleavage activity exhibited by Cas12a and Cas13a forms the foundation for their biosensing applications, enabling signal amplification through the cleavage of reporter molecules [19] [34].
Workflow Integration
The complete diagnostic workflow extends from sample collection to result interpretation, with sample preparation serving as the critical first step. Effective integration requires consideration of compatibility between extraction methods and downstream CRISPR detection, including buffer composition, volume requirements, and potential inhibition factors [50] [69].
Sample Processing Methodologies
Food Matrix Processing
Food fermentation samples present unique challenges due to their complex composition, which may include particulate matter, proteins, lipids, and carbohydrates that can inhibit downstream reactions. Effective processing methods must address these challenges while preserving target nucleic acids:
Liquid Fermentation Samples: For liquid samples such as beer, wine, or liquid fermentation broths, centrifugation at 12,000 × g for 10 minutes effectively concentrates microbial cells while removing particulate debris [69]. The resulting pellet can be resuspended in a minimal volume of appropriate buffer for subsequent extraction.
Solid and Semi-Solid Samples: Solid fermentation products (cheese, fermented meats, solid-state fermentation substrates) require additional processing. Aseptic homogenization in phosphate-buffered saline (PBS) using a 1:5 (w/v) sample to buffer ratio creates a uniform suspension [50]. Stomaching or bead beating effectively disrupts solid matrices and releases microbial cells.
Viscous Samples: Highly viscous samples such as yogurt or thick fermentation slurries benefit from dilution with specialized extraction buffers containing surfactants to reduce viscosity and improve extraction efficiency.
Microbial Enrichment and Concentration
In food fermentation samples where target microorganisms may be present in low abundance, enrichment strategies improve detection sensitivity:
Filtration: Membrane filtration (0.22-0.45 μm pore size) effectively concentrates bacterial cells from liquid samples, particularly useful for low-biomass samples like clarified fermentation products.
Differential Centrifugation: Sequential centrifugation at lower speeds (500 × g for 5 minutes) to remove food particles, followed by higher speeds (12,000 × g for 10 minutes) to pellet microbial cells, effectively separates microorganisms from matrix components.
Immunomagnetic Separation: Antibody-coated magnetic beads specific to target pathogens provide selective concentration from complex samples, significantly improving detection limits in CRISPR-based assays [50].
Nucleic Acid Extraction Strategies
Laboratory-Based Extraction Methods
For laboratory settings with access to specialized equipment, several extraction methods provide high-quality nucleic acids suitable for CRISPR detection:
Table 1: Comparison of Nucleic Acid Extraction Methods
| Method | Principle | Yield | Purity (A260/A280) | Processing Time | Cost per Sample | Compatibility with CRISPR |
|---|---|---|---|---|---|---|
| Spin Column | Silica-membrane binding with wash steps | High | 1.8-2.0 | 30-45 min | High | Excellent |
| Magnetic Beads | Magnetic silica particle binding | High | 1.7-2.0 | 20-30 min | Medium | Excellent |
| Liquid-Liquid | Organic phase separation | Medium | 1.6-1.9 | 60+ min | Low | Good (requires purification) |
| FTA Cards | Cell lysis and nucleic acid capture on card | Variable | N/A | 15-20 min | Very Low | Good |
Spin Column Technology: This method utilizes silica-based membranes in column format that bind nucleic acids in the presence of chaotropic salts. After binding, contaminants are removed through wash steps, and pure nucleic acids are eluted in low-salt buffers or water. This method consistently yields high-purity DNA and RNA suitable for CRISPR-based detection [50].
Magnetic Bead-Based Extraction: Superparamagnetic particles coated with silica provide a high-surface-area substrate for nucleic acid binding. This technology enables automation and high-throughput processing, with several commercial systems specifically validated for food matrix applications [69].
Simplified and Equipment-Free Methods
For field deployment or resource-limited settings, simplified extraction methods offer practical alternatives:
- FTA Cards: Flinders Technology Associates (FTA) cards contain chemicals that lyse microbial cells, denature proteins, and protect nucleic acids from degradation. The sample is applied directly to the card, where cellular components are separated, and nucleic acids are immobilized within the matrix [69]. For analysis, a small disc is punched from the card and washed, then used directly in amplification reactions without further purification.
Protocol: FTA Card-Based Nucleic Acid Extraction
- Apply 100-500 μL of liquid sample or homogenate to the FTA card surface
- Air dry the card completely at room temperature for 1-2 hours
- Punch a 2-3 mm disc from the sample area using a sterile disposable punch
- Transfer the disc to a microcentrifuge tube
- Wash twice with 200 μL FTA purification reagent (5 minutes per wash with gentle mixing)
- Wash once with 200 μL TE buffer (10 mM Tris-HCl, 0.1 mM EDTA, pH 8.0) for 5 minutes
- Remove wash buffer completely and air dry the disc at 56°C for 10 minutes or room temperature for 30 minutes
- Use the dried disc directly in downstream amplification reactions [69]
- Boiling Extraction: Simple heating of samples in alkaline buffers or water can release nucleic acids through thermal lysis. While this method provides limited purity, it is extremely rapid and low-cost. For CRISPR detection, a brief centrifugation step is recommended to remove particulate debris that may inhibit downstream reactions.
Integration with CRISPR-Based Detection
Direct Integration with Amplification-Free CRISPR
Amplification-free CRISPR detection approaches are particularly compatible with simplified extraction methods, as they minimize sample handling and reduce contamination risk:
Cas12a-Based DNA Detection: FTA card extracts containing bacterial DNA can be detected directly using Cas12a systems without additional purification. The trans-cleavage activity of Cas12a against ssDNA reporters provides measurable signals even at low target concentrations [68] [69].
Cas13a-Based RNA Detection: For RNA targets in fermentation monitoring, extraction methods that preserve RNA integrity are essential. FTA cards have demonstrated effectiveness in stabilizing RNA for subsequent detection with Cas13a-based systems [34].
Integration with Pre-Amplification CRISPR Methods
When maximum sensitivity is required, nucleic acid extraction is followed by isothermal amplification before CRISPR detection:
Protocol: Recombinase Polymerase Amplification (RPA) Integration
- Prepare RPA reaction mix according to manufacturer's instructions (typically 50 μL total volume)
- Add 1-5 μL of extracted nucleic acids or one FTA card disc directly to the reaction mixture
- Incubate at 37-42°C for 15-25 minutes
- Transfer 1-2 μL of amplified product to CRISPR detection reaction
- Perform CRISPR detection at 37°C with real-time or endpoint fluorescence measurement [19] [69]
This combined approach achieves exceptional sensitivity, with detection limits as low as 1 copy/μL for bacterial pathogens in food matrices [19].
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Research Reagents for Sample Preparation and CRISPR Detection
| Reagent/Material | Function | Application Notes |
|---|---|---|
| FTA Cards | Nucleic acid capture, storage, and purification | Enables equipment-free sample processing; compatible with complex food matrices [69] |
| EnGen Lba Cas12a | CRISPR effector protein for DNA detection | Recognizes T-rich PAM sequences; high trans-cleavage activity [19] [69] |
| TwistAmp Basic RPA Kit | Isothermal nucleic acid amplification | Enables sensitive detection at constant temperature; compatible with crude extracts [69] |
| Thioflavin T (ThT) | G-quadruplex binding fluorescent dye | Label-free reporter for CRISPR detection; significant cost savings over labeled probes [69] |
| Custom crRNAs | Target-specific guide RNAs | Programmable for different microbial targets; design complementary to conserved genomic regions [68] [34] |
| Magnetic Silica Beads | Nucleic acid purification | Enable high-throughput processing; effective with inhibitory food matrices [50] |
Troubleshooting and Optimization
Addressing Common Challenges
Inhibition of CRISPR Reactions: Complex food matrices may contain components that inhibit Cas enzyme activity. Dilution of extracted nucleic acids or the addition of enhancers such as BSA (0.1-0.5 mg/mL) can mitigate inhibition effects [50].
Low Nucleic Acid Yield: For samples with low microbial load, increasing starting sample volume or incorporating a brief enrichment culture step improves detection capability. Alternatively, larger FTA card discs (up to 6 mm) can be used to increase template amount.
Variable Extraction Efficiency: Implementation of internal controls, such as exogenous DNA spikes, monitors extraction efficiency and identifies processing failures. Consistent sample-to-buffer ratios and processing times improve reproducibility.
Quality Assessment
Rapid quality assessment methods ensure extracted nucleic acids are suitable for downstream CRISPR detection:
Spectrophotometric Analysis: When equipment is available, A260/A280 ratios between 1.8-2.0 indicate pure DNA preparations, while values around 2.0-2.2 suggest high-quality RNA.
Rapid Integrity Check: Amplification of a conserved gene target (e.g., 16S rRNA for bacteria) using a rapid PCR or RPA format verifies both nucleic acid quality and the absence of PCR inhibitors.
Effective sample preparation and nucleic acid extraction are fundamental to successful implementation of CRISPR-based biosensors in food fermentation research. The strategies presented here, particularly the integration of FTA card-based extraction with CRISPR detection, provide a streamlined pathway from sample to result that maintains analytical sensitivity while significantly simplifying processing requirements. As CRISPR technologies continue to evolve toward greater sensitivity and multiplexing capability, parallel advancements in sample preparation will ensure these powerful detection tools can be effectively deployed across diverse food fermentation applications, from routine quality control to investigation of contamination events. The protocols and methodologies detailed in this application note provide researchers with practical frameworks for implementing these integrated approaches in both laboratory and field settings.
The application of CRISPR-based biosensors for microbial detection in food fermentation research represents a transformative advancement in monitoring complex microbial communities. However, the accuracy of these platforms is critically dependent on overcoming two primary challenges: off-target effects and non-specific amplification. Off-target effects occur when CRISPR-Cas systems cleave non-target DNA or RNA sequences with partial complementarity to the guide RNA, potentially generating false-positive signals in biosensing applications [71]. Simultaneously, non-specific amplification in isothermal amplification methods like RPA and LAMP can produce spurious amplification products that activate CRISPR collateral activity, further compromising detection accuracy [68]. These challenges are particularly pronounced in food fermentation environments, where diverse microbial communities and complex food matrices create additional obstacles for precise detection. This application note provides a comprehensive framework of strategies, protocols, and reagents to enhance the specificity of CRISPR-based biosensing in food fermentation research.
Mechanisms of Off-Target Effects and Non-Specific Amplification
The collateral activity of Cas proteins, while fundamental to CRISPR-based detection, also represents a significant source of potential off-target effects. Different Cas proteins exhibit distinct off-target profiles, with Cas12a demonstrating preferential cleavage of non-target single-stranded DNA following target recognition, while Cas13 targets non-specific single-stranded RNA [68] [33]. This trans-cleavage activity, though harnessed for signal amplification in biosensors, can be triggered by sequences with partial complementarity, especially under suboptimal reaction conditions. Structural studies indicate that the REC2-Nuc interactions and specific loop structures within Cas12a orthologs significantly influence their cis and trans cleavage kinetics, contributing to variations in off-target propensity among different Cas protein variants [72].
The guide RNA-target sequence interaction represents another critical factor in off-target binding. While CRISPR systems typically require a protospacer adjacent motif (PAM) for initial target recognition, the sequence complementarity 5' of the PAM site exhibits greater tolerance for mismatches, particularly in high GC-content regions [68]. This flexibility, evolutionarily advantageous for bacterial immunity, becomes problematic for diagnostic applications where single-nucleotide discrimination is often essential. Furthermore, extended guide RNA sequences beyond the seed region can stabilize imperfect duplex formations through non-Watson-Crick base pairing, facilitating unintended activation of Cas nucleases [19].
Origins of Non-Specific Amplification in Sample Preparation
Isothermal amplification techniques, frequently coupled with CRISPR detection, are particularly susceptible to non-specific amplification due to their constant temperature operation and enzyme characteristics. Recombinase polymerase amplification (RPA) and loop-mediated isothermal amplification (LAMP) can generate false-positive amplification products through several mechanisms, including primer-dimer formations, mispriming events, and polymerase errors that create non-target amplicons capable of activating CRISPR systems [68]. These challenges are exacerbated in food fermentation samples, where high concentrations of background DNA from starter cultures or complex matrices may contain sequences with partial homology to target pathogens or spoilage organisms.
The composition of food samples introduces additional complexity, with polysaccharides, lipids, proteins, and other compounds potentially inhibiting enzymatic reactions or promoting non-specific interactions. Fermented products like yogurt, cheese, and fermented meats contain diverse microbial communities where target organisms may represent a small fraction of the total population, increasing the likelihood of non-target amplification [73] [18]. Moreover, metabolic byproducts including organic acids, exopolysaccharides, and bacteriocins can interfere with both amplification efficiency and CRISPR-Cas activity, further challenging detection specificity [18].
Table 1: Classification of Specificity Challenges in CRISPR-Based Microbial Detection
| Challenge Type | Underlying Mechanism | Impact on Detection |
|---|---|---|
| CRISPR Off-Target Effects | Partial complementarity between crRNA and non-target DNA/RNA | False positive signals from non-target activation |
| Cas12a Trans-Cleavage | Non-specific ssDNA degradation post-activation | Background signal generation independent of target |
| Cas13 Trans-Cleavage | Non-specific ssRNA degradation post-activation | Signal amplification from non-target RNA sources |
| Amplification Non-Specificity | Primer-dimer formation and mispriming in RPA/LAMP | Generation of non-target amplicons that activate Cas systems |
| Matrix Interference | Food components inhibiting enzymatic reactions | Reduced efficiency leading to compensatory over-amplification |
Strategic Approaches for Enhanced Specificity
CRISPR-Cas Protein Engineering and Selection
Strategic selection and engineering of Cas proteins represents a fundamental approach to enhancing detection specificity. The development of high-fidelity Cas variants through rational design and directed evolution has yielded proteins with reduced off-target propensity while maintaining robust on-target activity. Notably, the introduction of TadA8e mutants with modified residues (H52L/D53R) in adenine base editors has demonstrated significantly minimized RNA editing activity while preserving efficient on-target DNA editing, addressing a critical source of off-target effects in base editing applications [72]. These engineered variants enable more precise detection of single-nucleotide polymorphisms (SNPs) relevant for distinguishing between closely related microbial strains in fermentation communities.
The exploration of diverse Cas orthologs beyond the commonly used Cas9 and Cas12a has identified enzymes with inherently higher specificity profiles. Cas12f (formerly Cas14), despite its compact size, exhibits exceptional discrimination capability for short single-stranded DNA targets without requiring a PAM sequence, making it particularly suitable for detecting SNP markers in microbial communities [68] [33]. Similarly, the recently characterized CasΦ (Cas12j) demonstrates minimal off-target activity while maintaining robust detection sensitivity, offering an additional option for specific detection in complex samples [68]. For food fermentation applications requiring discrimination between closely related lactic acid bacteria strains, these high-specificity Cas variants provide critical advantages for precise microbial monitoring.
The engineering of Cas12a direct repeat (DR) mutants through systematic mutation of the direct repeat sequence enables more precise control over Cas12a activation, significantly improving base editing accuracy and reducing non-specific cleavage [72]. This crRNA engineering approach represents a programmable strategy for enhancing specificity without modifying the Cas protein itself, offering flexibility for different detection scenarios. Furthermore, the development of Cas9 nickase variants that generate single-strand breaks rather than double-strand breaks, particularly when employed in dual guide RNA configurations targeting opposite strands, demonstrates substantially reduced off-target effects while maintaining efficient on-target activity [71].
crRNA Design and Reaction Optimization
Sophisticated crRNA design strategies provide a powerful approach to enhancing specificity without requiring protein engineering. Bioinformatic tools for guide RNA selection should incorporate comprehensive off-target prediction algorithms that evaluate potential cross-reactivity with non-target sequences present in the food fermentation microbiome. The implementation of truncated guide RNAs with shorter spacer sequences (17-18 nt instead of 20 nt) has demonstrated improved specificity by reducing tolerance to mismatches, particularly in regions distal to the PAM sequence [19]. For discrimination of single-nucleotide variants (SNVs) in highly conserved microbial genes, the strategic positioning of the mismatch at specific positions within the guide RNA seed region (typically positions 8-15) can dramatically enhance discriminatory capability.
Reaction condition optimization represents a complementary approach to maximizing specificity. Adjustment of Mg²⁺ concentration and elevated reaction temperatures can significantly enhance the stringency of Cas protein activation, preferentially favoring perfectly matched target sequences over those with mismatches [19]. The proPE system, which utilizes a second non-cleaving sgRNA to position the prime editing machinery in closer proximity to the edit site, demonstrates 6.2-fold enhancement in editing efficiency for previously challenging targets while reducing the optimization requirements compared to standard prime editing approaches [72]. For food fermentation applications, incorporating blocking oligonucleotides that bind to and protect highly similar non-target sequences present in the sample matrix can further prevent off-target activation.
Table 2: Strategic Approaches for Mitigating Specificity Challenges
| Strategy Category | Specific Approach | Mechanism of Action | Applicable Cas Systems |
|---|---|---|---|
| Protein Engineering | High-fidelity Cas variants (TadA8e mutants) | Reduced RNA off-target editing while maintaining DNA activity | ABE, CBE |
| Ortholog Selection | Cas12f (Cas14) utilization | Enhanced discrimination for short ssDNA targets without PAM requirement | Cas12f |
| crRNA Design | Truncated guide RNAs (tru-gRNAs) | Reduced mismatch tolerance through shorter complementarity regions | Cas9, Cas12a, Cas13 |
| Reaction Optimization | Elevated temperature and adjusted Mg²⁺ | Enhanced stringency of target recognition | All Cas systems |
| Workflow Innovation | Amplification-free CRISPR detection | Elimination of amplification-derived non-specific products | Cas12a, Cas13 |
Amplification-Free CRISPR Detection Platforms
The development of amplification-free CRISPR detection strategies directly addresses the challenge of non-specific amplification by eliminating the pre-amplification step entirely. These approaches leverage the intrinsic sensitivity of Cas proteins combined with enhanced signal detection systems to achieve target detection without amplification. The CRISPR-Cas13a amplification-free platform for SARS-CoV-2 detection demonstrates a remarkable sensitivity of 470 aM within 30 minutes, establishing the feasibility of direct detection without amplification [68]. For food fermentation applications where rapid monitoring is prioritized over ultra-sensitive detection, such platforms effectively eliminate amplification-derived false positives while providing sufficient sensitivity for quality control purposes.
Advanced signal enhancement methodologies enable the practical implementation of amplification-free detection. Cascade CRISPR systems incorporate multiple Cas proteins in sequential activation cascades, significantly amplifying the detection signal without nucleic acid amplification [68]. Integrated sensor technologies including graphene field-effect transistors (gFET), electrochemical (ECL) sensors, and surface-enhanced Raman spectroscopy (SERS) platforms transduce CRISPR activation into measurable physical signals with attomolar to femtomolar sensitivity, enabling direct detection of microbial targets [68] [33]. The digital droplet CRISPR platform partitions samples into numerous individual reactions, enabling absolute quantification of target molecules through Poisson distribution analysis without pre-amplification [68].
For food fermentation research, amplification-free detection offers particular advantages for monitoring dominant microbial populations where extreme sensitivity is less critical than specificity and speed. The implementation of microfluidic integration with amplification-free CRISPR detection enables automated, high-throughput analysis of fermentation samples, facilitating real-time monitoring of microbial dynamics throughout the fermentation process [19]. Furthermore, the reduced reagent requirements and simplified workflow of amplification-free approaches enhance their suitability for point-of-use testing in food production facilities where sophisticated laboratory equipment may be unavailable.
Experimental Protocols for Specificity Enhancement
Protocol: High-Specificity crRNA Design and Validation
Principle: This protocol outlines a comprehensive approach for designing and validating highly specific crRNAs for microbial detection in food fermentation matrices, incorporating bioinformatic screening and experimental confirmation to minimize off-target effects.
Materials:
- Target microbial sequences from databases (NCBI, RDP, SILVA)
- Background genome database of fermentation microbiota
- crRNA design software (CHOPCHOP, CRISPRscan, or custom tools)
- In vitro transcription kit for crRNA synthesis
- Purified Cas protein (Cas12a, Cas9, or Cas13 as required)
- Synthetic target and off-target DNA/RNA templates
- Fluorescent reporter probes (ssDNA-FQ for Cas12a, RNA for Cas13)
Procedure:
- Target Sequence Selection:
- Identify unique genomic regions in target microorganisms with minimal homology to non-target species in the fermentation community.
- For bacterial detection, target single-copy genes with strain-specific variable regions (e.g., 16S rRNA V regions, housekeeping genes).
- Avoid sequences with high GC content (>70%) or repetitive elements that promote non-specific binding.
Bioinformatic crRNA Design:
- Generate candidate crRNA spacers of 20-22 nt complementary to the target region.
- Screen all candidates against a comprehensive database of fermentation-relevant microorganisms using BLASTN or specialized CRISPR off-target prediction tools.
- Select crRNAs with ≥3 mismatches to all non-target sequences, prioritizing mismatches in the seed region (positions 3-10 proximal to PAM).
- For ultimate specificity, design truncated crRNAs (17-18 nt) while verifying maintained on-target efficiency.
Experimental Validation:
- Synthesize selected crRNAs using in vitro transcription with standardized purification.
- Test crRNA efficiency against synthetic target templates in serial dilutions to establish limit of detection.
- Challenge each crRNA with synthetic off-target templates containing 1-3 mismatches at different positions.
- Quantify signal generation for both target and off-target sequences using fluorescence kinetics.
- Select crRNAs demonstrating at least 100-fold signal discrimination between target and closest off-target sequence.
Troubleshooting:
- Low on-target signal: Extend crRNA length by 1-2 nucleotides or adjust reaction temperature.
- High off-target signal: Introduce additional mutations into crRNA direct repeat region or implement dual crRNA approach requiring simultaneous recognition.
- Variable performance across replicates: Standardize crRNA purification and storage conditions to maintain consistency.
Protocol: Amplification-Free CRISPR Detection with Electrochemical Readout
Principle: This protocol describes an amplification-free detection approach combining CRISPR activation with electrochemical signal transduction, eliminating amplification-derived false positives while maintaining high sensitivity for microbial targets in food fermentation samples.
Materials:
- Cas12a or Cas13 protein (purified, commercial or in-house)
- Specific crRNA designed per Protocol 4.1
- Screen-printed gold electrodes or graphene field-effect transistors (gFET)
- Electrochemical reporter (methylene blue-labeled ssDNA for Cas12a, RNA for Cas13)
- Potentiostat for electrochemical measurements
- Food sample pre-treatment reagents (lysozyme, proteinase K, homogenization buffer)
- Nucleic acid extraction and purification kit
Procedure:
- Sample Preparation:
- Homogenize 1g of food fermentation sample in 10mL of sterile phosphate-buffered saline (PBS).
- Centrifuge at 5,000 × g for 10 minutes to collect microbial cells.
- Resuspend pellet in lysis buffer containing lysozyme (10mg/mL) and proteinase K (0.1mg/mL).
- Incubate at 56°C for 30 minutes followed by heat inactivation at 95°C for 5 minutes.
- Purify nucleic acids using silica column-based extraction, eluting in 50μL nuclease-free water.
Sensor Preparation:
- Functionalize electrodes with thiolated capture probes complementary to a region adjacent to the CRISPR target site.
- Block non-specific binding sites with 1% BSA in PBS for 1 hour at room temperature.
- Assemble CRISPR detection complex by pre-incubating Cas protein (50nM) with crRNA (60nM) in reaction buffer for 10 minutes at 25°C.
Detection Reaction:
- Mix 10μL of extracted nucleic acids with 30μL of pre-assembled CRISPR complex.
- Incubate at 37°C for 15 minutes to allow target recognition and trans-cleavage activation.
- Apply the reaction mixture to the functionalized electrode and incubate for 5 minutes.
- Measure electrochemical signal (differential pulse voltammetry or electrochemical impedance spectroscopy).
Data Analysis:
- Calculate signal-to-background ratio by comparing sample signals to negative controls.
- Quantify target concentration using a standard curve generated with synthetic targets.
- Establish threshold values for positive detection based on negative control population (mean + 3 standard deviations).
Troubleshooting:
- High background signal: Increase stringency of electrode washing or optimize blocking conditions.
- Low sensitivity: Extend reaction incubation time or increase Cas protein concentration within optimal range.
- Matrix interference: Implement additional sample clean-up steps or sample dilution in complex matrices.
Research Reagent Solutions
Table 3: Essential Reagents for Specific CRISPR-Based Detection in Food Fermentation Research
| Reagent Category | Specific Product | Function & Application | Specificity Considerations |
|---|---|---|---|
| High-Fidelity Cas Proteins | Alt-R S.p. HiFi Cas9 | Reduced off-target editing while maintaining on-target activity | Engineered variant with point mutations for enhanced specificity |
| Specialized Cas Variants | Cas12f (Cas14) | Ultra-specific ssDNA targeting without PAM requirement | Compact size with inherent high discrimination capability |
| crRNA Design Tools | CHOPCHOP web tool | Bioinformatic guide RNA design with off-target prediction | Incorporates comprehensive food microbe databases |
| Detection Reporters | ssDNA-FQ reporters (FAM-TTATT-BHQ1) | Fluorescent signal generation upon Cas activation | Optimal length and sequence to minimize background cleavage |
| Amplification Reagents | Alt-R HDR Enhancer Protein | Improved homology-directed repair efficiency | Reduces need for excessive amplification that promotes non-specificity |
| Sample Preparation | Lysozyme & proteinase K | Microbial lysis and nucleic acid liberation | Efficient extraction reduces amplification requirements |
| Signal Detection | Screen-printed gold electrodes | Electrochemical transduction of CRISPR activation | Enables amplification-free detection approaches |
Workflow Visualization
Specificity Enhancement Workflow: This workflow illustrates the comprehensive approach to enhancing specificity in CRISPR-based detection for food fermentation research, incorporating both amplification-based and amplification-free pathways with iterative optimization.
Off-Target Mechanisms and Mitigation: This diagram illustrates the primary mechanisms of off-target effects in CRISPR-based detection and the corresponding mitigation strategies that enhance specificity for accurate microbial detection in food fermentation research.
The integration of specificity-enhancing strategies throughout the CRISPR-based detection workflow is essential for accurate microbial monitoring in complex food fermentation environments. The complementary approaches of Cas protein engineering, sophisticated crRNA design, reaction optimization, and amplification-free detection provide a comprehensive toolkit for addressing both off-target effects and non-specific amplification. As CRISPR biosensing continues to evolve, emerging technologies including artificial intelligence-guided guide RNA design, miniaturized Cas variants with enhanced discrimination capabilities, and multi-target detection arrays will further advance the specificity and applicability of these platforms for food fermentation research [59] [71].
The implementation of the protocols and strategies outlined in this application note will enable researchers to achieve unprecedented specificity in monitoring starter cultures, detecting contamination events, and profiling microbial community dynamics throughout fermentation processes. By systematically addressing the fundamental challenges of off-target effects and non-specific amplification, CRISPR-based biosensors can fulfill their potential as robust, reliable tools for advancing food fermentation science and ensuring product quality and safety.
Lyophilization and Room-Temperature Storage for Enhanced Portability and Shelf-Life
Within food fermentation research, the demand for robust, on-site diagnostic tools is paramount. CRISPR-based biosensors have emerged as a powerful technology for the specific and sensitive detection of microbial contaminants and the monitoring of starter cultures [6] [18]. However, the transition of these biosensors from controlled laboratory settings to real-world applications in production facilities or field settings is hindered by their reliance on cold-chain storage and handling. Lyophilization, or freeze-drying, presents a strategic solution to this challenge. By removing water under vacuum, biochemical reagents can be stabilized into a solid, dry state, dramatically enhancing their shelf life at ambient temperatures and enabling easy transport [74]. This Application Note provides detailed protocols and data for the lyophilization and room-temperature storage of CRISPR-based biosensing platforms, specifically framed for their application in microbial detection within food fermentation research.
Quantitative Data on Stability and Performance
The following tables summarize key performance metrics from studies on lyophilized biological systems, highlighting the enhancements in stability and functionality critical for deploying CRISPR-based biosensors in non-laboratory settings.
Table 1: Comparative Stability of Lyophilized vs. Liquid Reagent Formats
| Reagent System | Storage Condition | Storage Duration | Residual Activity | Key Findings |
|---|---|---|---|---|
| Cell-Free Protein Synthesis (CFPS) [75] | Room Temperature | 2 weeks | ~35% (No supplements) | Liquid extracts lose all activity at 23°C within two weeks. |
| Lyophilized CFPS [75] | Room Temperature | 2 weeks | ~79% (With supplements) | Lyophilization alone significantly improves stability over liquid forms. |
| Lyophilized CFPS (DoE-Optimized) [75] | Room Temperature | 1 month | ~100% | An optimized combination of stabilizers (PEG, trehalose, trimethylglycine) enabled full preservation. |
| Lyophilized CRISPR Assay (SARS-CoV-2) [76] | Not Specified | Not Specified | 100% (Sensitivity), 99.05% (Specificity) | The lyophilized kit showed nearly perfect concordance (Kappa=0.991) with standard RT-qPCR. |
Table 2: Efficacy of Lyophilized CRISPR-Cas12 Assay for Pathogen Detection
| Evaluation Parameter | Performance Metric | Details / Implications |
|---|---|---|
| Clinical Sensitivity [76] | 100% (105/105 samples) | Correctly identified all positive samples previously confirmed by RT-qPCR. |
| Clinical Specificity [76] | 99.05% (104/105 samples) | Correctly identified almost all negative samples, demonstrating high specificity. |
| Dynamic Range [76] | Ct 11.45 - 36.90; 2.5-100 copies/µL | Suitable for detecting a wide range of target concentrations, from high to low viral loads. |
| Cross-Reactivity [76] | No cross-reaction observed | Assay was specific to the target pathogen amidst other common respiratory pathogens. |
Experimental Protocols
Lyophilization Protocol for CRISPR-Cas Biosensing Reactions
This protocol describes the process for creating stable, lyophilized pellets of CRISPR-Cas detection reagents, suitable for detecting microbial targets like Listeria monocytogenes or Salmonella spp. in food fermentation samples [76] [74].
- Step 1: Reagent Assembly. On ice, combine the following core components to form a master mix:
- CRISPR-Cas Enzyme: Cas12a (for DNA targets) or Cas13a (for RNA targets). Use a final concentration of 50-100 nM.
- Guide RNA (crRNA): Design a species-specific crRNA targeting a unique genomic sequence of the foodborne pathogen of interest (e.g., hlyA for L. monocytogenes). Use a final concentration of 50-100 nM.
- Fluorescent Reporter: A quenched single-stranded DNA (for Cas12a) or RNA (for Cas13a) reporter molecule (e.g., FAM-TTATT-BHQ1). Use a final concentration of 500 nM - 1 µM.
- Reaction Buffer: Provide appropriate salts (e.g., MgCl₂, KGlutamate), buffering agents (e.g., Tris-HCl, HEPES), and DTT for nuclease activity and stability.
- Stabilizing Excipients: Add the optimized combination of stabilizers determined from screening (e.g., 100 mM Trehalose, 2% w/v PEG-8000, 500 mM Trimethylglycine). These act as cryoprotectants and lyoprotectants [75].
- Step 2: Aliquoting and Flash-Freezing. Dispense the assembled master mix into the desired reaction vessels (e.g., PCR tubes or microtiter plates). Immediately after aliquoting, flash-freeze the samples by submerging them in liquid nitrogen (-196 °C) for 60-90 seconds to ensure rapid and uniform freezing [74].
- Step 3: Primary Drying (Lyophilization). Quickly transfer the frozen samples to a pre-cooled shelf in a freeze-drier. Apply a high vacuum (e.g., < 100 mTorr). The primary drying phase, where ice sublimates, typically takes 3-6 hours. The duration must be optimized based on aliquot volume and equipment.
- Step 4: Secondary Drying (Desorption). After primary drying, the shelf temperature may be gradually increased (e.g., to 25 °C) for a secondary drying phase of 2-4 hours. This step removes bound water, further stabilizing the reagents for long-term storage.
- Step 5: Sealing and Storage. Once the cycle is complete, backfill the chamber with an inert gas (e.g., argon or nitrogen) if available, and hermetically seal the containers under vacuum or inert atmosphere. Store the lyophilized pellets at room temperature, protected from light and moisture.
Protocol for Enhancing Stability via a Design of Experiments (DoE) Approach
This methodology outlines a systematic, minimalistic DoE to identify optimal stabilizer combinations for maximizing the room-temperature shelf life of lyophilized biosensors [75].
- Step 1: Initial Supplement Screening. Select a panel of candidate stabilizers with known protective mechanisms:
- Sugars: Trehalose, Sucrose (water replacement, vitrification).
- Crowding Agents: PEG 6000, PEG 8000 (molecular crowding, stabilization).
- Osmolytes: Trimethylglycine (aka Betaine, stabilizes protein structure).
- Step 2: High-Throughput Activity Assay. Reconstitute lyophilized reactions containing different single stabilizers and their combinations. Use a kinetic fluorescence assay (e.g., with a reporter like sfGFP or the fluorescent CRISPR reporter) to measure functional activity. Compare this to a freshly prepared liquid control (100% activity) and a negative control (0% activity).
- Step 3: Minimalistic DoE Design. Based on initial screening, choose the top 2-3 performing stabilizers and design a simple factorial experiment (e.g., a 2^3 full factorial design) to test their interactions. The factors are the stabilizers, and the levels are a low and high concentration for each.
- Step 4: Stability Testing. Lyophilize the DoE formulations and store them at room temperature and accelerated stability conditions (e.g., 37 °C). At predetermined time points (e.g., 1 week, 2 weeks, 1 month), reconstitute samples and measure residual activity.
- Step 5: Data Analysis and Optimization. Analyze the activity data to determine the main effects and interaction effects of each stabilizer. Identify the specific combination and concentrations that yield 100% preservation of activity over the target shelf life [75].
The Scientist's Toolkit
Table 3: Essential Research Reagent Solutions for Lyophilization
| Item | Function / Role in Lyophilization |
|---|---|
| Trehalose | A non-reducing disaccharide that acts as a superior lyoprotectant; forms an amorphous glassy matrix that replaces water and protects protein structure during drying and storage [75]. |
| PEG (Polyethylene Glycol) | A molecular crowding agent that stabilizes proteins and can improve folding; also contributes to the stabilization of the lyophilized cake structure [75]. |
| Trimethylglycine (Betaine) | An osmolyte that stabilizes proteins against denaturation caused by stress conditions such as freezing and drying [75]. |
| Lyophilized CRISPR Kit | A pre-formulated, ready-to-use kit that integrates reverse transcription, isothermal amplification, and CRISPR detection in a single lyophilized pellet, enabling sensitive and specific pathogen detection in resource-limited settings [76]. |
| Nuclease-Free Water | The essential reagent for reconstituting lyophilized pellets; must be free of nucleases to prevent degradation of CRISPR reagents (gRNA, reporters) and target nucleic acids. |
Workflow and Signaling Pathways
The following diagram illustrates the integrated workflow of a lyophilized CRISPR-based biosensor, from sample preparation to final detection, highlighting the key molecular signaling events.
Multiplexing Strategies for Simultaneous Detection of Multiple Microbial Targets
In food fermentation research, the microbial community plays a pivotal role in determining product yield, quality, and safety [77]. Profiling this complex ecosystem requires detection methods that can identify multiple pathogens or spoilage organisms simultaneously. Multiplex detection addresses this need by enabling the parallel analysis of several targets in a single reaction, saving time, reducing costs, and providing a comprehensive view of the microbial landscape [7] [78].
Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR)-based biosensors have emerged as powerful tools for nucleic acid detection due to their high specificity, sensitivity, and programmability [79] [32]. This application note details practical strategies for designing CRISPR-based biosensors for the simultaneous detection of multiple microbial targets relevant to food fermentation, including pathogenic bacteria such as Escherichia coli, Salmonella enterica, Staphylococcus aureus, and Listeria monocytogenes [7] [78].
CRISPR System Selection for Multiplexing
The foundation of a successful multiplex assay lies in selecting the appropriate CRISPR-Cas system. Class 2 systems, which utilize a single effector protein, are most suitable for diagnostic applications due to their simplicity [79]. Their distinct cleavage activities and substrate preferences, summarized in Table 1, provide a toolkit for designing multiplexed assays.
Table 1: Characteristics of Common Class 2 CRISPR-Cas Effector Proteins for Biosensing
| Effector Protein | CRISPR Type | Target Molecule | trans-cleavage Activity | PAM/PFS Requirement | Guide RNA |
|---|---|---|---|---|---|
| Cas9 | Type II | dsDNA | No | 5'-NGG PAM | tracrRNA:crRNA (often fused as sgRNA) |
| Cas12a | Type V | dsDNA, ssDNA | Yes, ssDNA | 5'-TTTN PAM | crRNA |
| Cas13a | Type VI | ssRNA | Yes, ssRNA | 3' non-G PFS | crRNA |
| Cas14a | Type V | ssDNA | Yes, ssDNA | Not for ssDNA; 5' T-rich for dsDNA | tracrRNA:crRNA |
The trans-cleavage activity—the nonspecific degradation of surrounding nucleic acids after target recognition—is key for signal amplification in biosensing [7] [79]. Cas12, Cas13, and Cas14 possess this activity, making them particularly valuable. For multiplexing, Cas12 and Cas13 are highly advantageous. Their single crRNA guide structure simplifies assay design, and their different target preferences (DNA for Cas12, RNA for Cas13) enable orthogonal detection pathways in a single reaction [79] [22]. For instance, Cas13 can be deployed to detect RNA viruses or gene expression markers, while Cas12 targets DNA-based pathogens [79].
Multiplexing Strategy 1: Spatial Separation on Solid Supports
This strategy physically separates different detection reactions within a single device, typically a microfluidic chip or a paper-based analytical device (μPAD) [78] [80]. Each reaction chamber is pre-loaded with a unique CRISPR system programmed to detect a specific target.
Figure 1: Workflow for spatially separated multiplex detection using a microfluidic device.
Experimental Protocol
Title: Multiplexed Detection of Salmonella spp. and Listeria monocytogenes via Spatially Resolved CRISPR on a Microfluidic Chip.
Principle: The sample is split and directed into separate microfluidic chambers, each containing a CRISPR-Cas system programmed with a unique crRNA to detect a specific pathogen. Target recognition activates trans-cleavage, and the signal is read via a lateral flow strip [78] [80].
Materials:
- Reagents:
- LbCas12a or LwaCas13a protein (commercial sources, e.g., New England Biolabs).
- Custom crRNAs targeting Salmonella invA gene and Listeria hlyA gene (synthesized, HPLC-purified).
- Fluorescent or FQ-reporters (ssDNA for Cas12a, ssRNA for Cas13a).
- Isothermal amplification reagents (e.g., RPA or LAMP kits).
- Nuclease-free water.
- Equipment:
- Custom microfluidic chip with ≥2 reaction chambers.
- Micropipettes.
- Temperature control block or incubator (37-42°C).
- Fluorescence reader or lateral flow strip reader.
Procedure:
- Chip Priming: Pipette 15 µL of the pre-mixed CRISPR detection mix into each chamber of the dry microfluidic chip.
- Chamber 1 (Salmonella): 50 nM LbCas12a, 75 nM invA-specific crRNA, 2 µM ssDNA-FQ reporter.
- Chamber 2 (Listeria): 50 nM LbCas12a, 75 nM hlyA-specific crRNA, 2 µM ssDNA-FQ reporter.
Sample Loading: Introduce 10 µL of the amplified or extracted nucleic acid sample into the common inlet port. Allow capillary action or applied pressure to distribute the sample into each chamber.
Incubation: Seal the chip and incubate at 37°C for 15-20 minutes.
Signal Detection: Visualize fluorescence using a handheld blue light transilluminator or quantify with a microplate reader. For lateral flow readout, add the reaction mixture to the strip, and results will appear within 5 minutes.
Troubleshooting:
- Low Signal: Ensure crRNAs are specific and have minimal secondary structure. Optimize Cas protein and crRNA concentrations.
- Cross-Talk Between Chambers: Verify the microfluidic design prevents leakage between chambers.
Multiplexing Strategy 2: Sequential and Orthogonal Assays
This approach runs multiple detection reactions in the same tube by leveraging the unique properties of different CRISPR-Cas systems or by temporally separating signal readouts [79] [22]. A prominent method uses the orthogonal DNA (Cas12) and RNA (Cas13) targeting capabilities.
Figure 2: Logical workflow for a one-pot, orthogonal multiplex assay using Cas12 and Cas13.
Experimental Protocol
Title: One-Pot Orthogonal Detection of E. coli O157:H7 (eaeA gene) and a Model RNA Virus using Cas12 and Cas13.
Principle: A single reaction tube contains both Cas12 and Cas13 systems with their respective crRNAs and spectrally distinct reporters. DNA targets activate Cas12 to cleave one reporter, while RNA targets activate Cas13 to cleave another, allowing for multiplexed detection via a single fluorescence readout step [79] [22].
Materials:
- Reagents:
- LbCas12a protein.
- LwCas13a protein.
- crRNA targeting E. coli eaeA gene.
- crRNA targeting a conserved region of the RNA virus genome (e.g., Norovirus).
- ssDNA-FAM Reporter (e.g., 5'-FAM-TTATT-3IABkFQ-3').
- ssRNA-HEX Reporter (e.g., 5'-HEX-UU-3IABkFQ-3').
- NEBuffer r2.1 or a suitable commercial CRISPR reaction buffer.
Procedure:
- Reaction Setup: Prepare a master mix on ice containing:
- 1x NEBuffer r2.1
- 50 nM LbCas12a
- 50 nM LwCas13a
- 75 nM of each crRNA (eaeA and viral)
- 2 µM ssDNA-FAM Reporter
- 2 µM ssRNA-HEX Reporter
- Nuclease-free water to 23 µL
Initiation: Add 2 µL of the extracted nucleic acid sample (containing both DNA and RNA) to the master mix, bringing the total volume to 25 µL. Pipette to mix.
Incubation and Detection:
- Incubate the reaction at 37°C for 30-60 minutes in a real-time PCR machine or a fluorescence plate reader.
- Monitor fluorescence in the FAM channel (excitation/emission ~485/535 nm) for Cas12 activity and the HEX/JOE channel (excitation/emission ~535/556 nm) for Cas13 activity.
Data Analysis: A positive result for E. coli is indicated by a significant increase in FAM fluorescence over a no-template control. A positive result for the RNA virus is indicated by a significant increase in HEX fluorescence.
The Scientist's Toolkit: Essential Research Reagent Solutions
Successful implementation of multiplex CRISPR assays relies on key reagents and materials. Table 2 lists essential components and their functions.
Table 2: Key Research Reagent Solutions for CRISPR-based Multiplex Detection
| Reagent/Material | Function/Description | Example Application in Protocol |
|---|---|---|
| Cas12a Protein (LbCas12a, AsCas12a) | RNA-guided DNA nuclease; provides target-specific cis-cleavage and non-specific ssDNA trans-cleavage. | Core enzyme for detecting DNA targets from bacterial pathogens like Salmonella and Listeria. |
| Cas13a Protein (LwCas13a) | RNA-guided RNA nuclease; provides target-specific cis-cleavage and non-specific ssRNA trans-cleavage. | Core enzyme for detecting RNA targets from viruses or for gene expression analysis in orthogonal assays. |
| crRNA (CRISPR RNA) | Short, customizable RNA that guides the Cas protein to its complementary nucleic acid target. | Designed to be unique for each microbial target (e.g., invA for Salmonella, hlyA for Listeria). |
| Fluorophore-Quencher (FQ) Reporter | Single-stranded DNA or RNA oligonucleotide with a fluorophore and quencher; cleavage separates the pair, generating fluorescence. | ssDNA-FQ reporter for Cas12; ssRNA-FQ reporter for Cas13. Used for real-time signal detection. |
| Isothermal Amplification Reagents (RPA/LAMP) | Enzymatic kits for amplifying target nucleic acids at a constant temperature, enhancing detection sensitivity. | Used for pre-amplifying target genes from microbial samples prior to CRISPR detection to achieve attomolar sensitivity. |
| Lateral Flow Strips | Disposable nitrocellulose strips for visual detection; often used with biotin- and FAM-labeled reporters. | Provides a low-cost, equipment-free readout for field-deployable assays. |
| Microfluidic Chip | Device with micro-scale channels and chambers that fluidically manage multiple parallel reactions. | Platform for spatial separation multiplexing, enabling several tests to be run from a single sample input. |
CRISPR-based biosensors offer a versatile and powerful platform for the simultaneous detection of multiple microbial targets, which is crucial for monitoring the complex ecosystems of food fermentations. The strategies outlined here—spatial separation and orthogonal assay design—provide researchers with practical frameworks to develop multiplexed tests. The exceptional specificity and sensitivity of CRISPR-Cas systems, combined with these multiplexing capabilities, pave the way for advanced diagnostics that can ensure food safety, optimize fermentation processes, and accelerate research and development.
Automation and One-Pot Assay Designs for Simplified Workflows and High-Throughput
The integration of automation and one-pot assay designs is revolutionizing the application of CRISPR-based biosensors in microbial detection for food fermentation research. These advancements directly address critical challenges in traditional methods, including labor-intensive workflows, lengthy time-to-results, and risks of cross-contamination from multi-step procedures [18] [26]. One-pot assays consolidate nucleic acid amplification and CRISPR detection into a single reaction vessel, significantly simplifying operational workflows [81] [82]. When combined with automated platforms, these systems enable high-throughput screening and provide exceptional reproducibility, making them ideally suited for monitoring dynamic microbial communities in fermentation processes such as those in dairy, meat, and traditional products like Baijiu [18] [83]. The evolution toward these integrated systems represents a significant step in bridging the gap between traditional fermentation craftsmanship and modern Industry 4.0 capabilities, ensuring better control over product consistency, safety, and quality [26].
Key One-Pot Assay Strategies and Their Mechanisms
Thermally Regulated Asynchronous CRISPR-Enhanced (TRACE) Assay
The TRACE assay utilizes temperature to physically separate the amplification and detection phases within a single tube. This method employs Cas12b and a complementary ssRNA blocker to prevent premature cleavage of amplicons during the initial amplification stage. The process begins with a low-temperature phase (37°C) where recombinase polymerase amplification (RPA) occurs, while the ssRNA blocker inhibits Cas12b activity. This is followed by a higher-temperature phase (60°C) where the blocker dissociates, allowing activated Cas12b to cleave reporter molecules [81]. This thermal segregation enables the TRACE assay to achieve a limit of detection (LoD) as low as 2.5 copies/test for Monkeypox virus, matching the sensitivity of traditional two-step methods while maintaining a one-pot format. The entire process can be completed within 11-40 minutes, making it exceptionally rapid [81].
Integrated Microfluidic One-Pot Systems
Microfluidic technologies have advanced one-pot assays by enabling high-throughput multiplexing in a fully automated format. The hMC-CRISPR platform integrates RAA amplification with CRISPR/Cas13a detection on a centrifugal microfluidic chip. This system uses T7 RNA polymerase to create a natural separation between DNA amplification and Cas13a's RNA-targeting activity, allowing all reagents to be pre-loaded into the chip [84]. The design automatically manages sample distribution, mixing, amplification, and data reading within closed microfluidic structures, eliminating aerosol contamination risks. This approach has demonstrated an impressive attomolar (aM) level LoD for pathogenic Listeria species and can process eight samples simultaneously in about 60 minutes [84].
Table 1: Comparison of Key One-Pot CRISPR Assay Platforms
| Assay Platform | CRISPR System | Amplification Method | Key Feature | Reported LoD | Time |
|---|---|---|---|---|---|
| TRACE [81] | Cas12b + ssRNA blocker | RPA | Thermally segregated reactions | 2.5 copies/test | 11-40 min |
| hMC-CRISPR [84] | Cas13a | RAA | Microfluidic multiplexing | aM level | ~60 min |
| AIOD-CRISPR [82] | Cas12a (dual gRNA) | RPA | Single-step visual detection | 2 copies/μL (RNA) | ~30 min |
| One-pot Cas12a-RPA [82] | Cas12a | RPA | DoE-optimized | 0.5 copies/μL (DNA) | ~30 min |
Optimization Strategies for One-Pot Assays
Achieving optimal performance in one-pot assays requires careful balancing of reaction components. Statistical Design of Experiments (DoE) has proven invaluable for optimizing the numerous variables in these complex systems. Through DoE, researchers discovered that adding reverse transcription buffer and RNase inhibitor – components often omitted in one-pot reactions – significantly improved performance for SARS-CoV-2 detection [82]. DoE also revealed that template-specific optimization is essential, as optimal conditions differ between RNA and DNA templates. This approach enabled detection of 2 copies/μL of SARS-CoV-2 RNA and 0.5 copies/μL of DNA fragments, among the lowest copy numbers detected using CRISPR/Cas12 technology [82].
Automation Platforms for High-Throughput CRISPR Workflows
Modular Robotic Systems
The StemCellFactory represents an advanced approach to automated CRISPR workflow management. This modular robotic system integrates a 4D-Nucleofector with a 96-well shuttle device for standardized genome editing operations. The platform connects various instruments via a robotic arm that handles cell cultures in multi-titer plates, managed by the COPE software system for process control, data tracking, and error handling [85]. This system has demonstrated indel rates up to 98% in human induced pluripotent stem cells (hiPSCs), comparable to manual methods while providing superior scalability and reproducibility. The integration of deep learning algorithms enables automated detection of cell differentiation states and confluence-based splitting procedures, accounting for clone-dependent growth variations [85].
Microfluidic Automation Systems
Microfluidic platforms provide liquid handling automation at a miniature scale. The hMC-CRISPR system exemplifies this approach by embedding all detection reagents within a centrifugal microfluidic chip. After sample introduction, the chip automatically executes sample distribution, mixing, amplification, and fluorescence detection without manual intervention [84]. This closed-system design eliminates cross-contamination risks while enabling multiplexed detection of multiple targets. The system's compact footprint and minimal reagent requirements (reactions as small as 5 μL) make it ideal for resource-limited settings while maintaining high-throughput capabilities of up to 32 parallel reactions [84].
Table 2: Research Reagent Solutions for Automated CRISPR Workflows
| Reagent Category | Specific Examples | Function in Workflow | Application Notes |
|---|---|---|---|
| Cas Proteins | LbCas12a, LwaCas13a, Cas12b | Target recognition and trans-cleavage | Cas12b used in TRACE for broader temperature range [81] |
| Amplification Systems | RPA, RAA, LAMP | Isothermal nucleic acid amplification | RPA compatible with 37-42°C range [82] |
| Reporters | ssDNA-FQ, RNA-FAM-BHQ1 | Fluorescent signal generation | Poly T reporters optimal for Cas12b [81] |
| gRNA Design | crRNA, tracrRNA, dual gRNAs | Target-specific recognition | Dual gRNAs enhance signal intensity [82] |
| Enhancers | Electroporation enhancer, RNase inhibitor | Improve efficiency and stability | Critical for one-pot assay sensitivity [82] [85] |
| Buffers | NEBuffer 2.1, P3 Nucleofection Buffer | Maintain optimal reaction conditions | Reverse transcription buffer boosts one-pot performance [82] |
Application Notes for Food Fermentation Research
Monitoring Functional Microbes in Fermentation
CRISPR-based biosensors have been successfully applied to monitor key microorganisms in food fermentation processes. A CRISPR/Cas12a-coupled gold nanoparticle visual biosensor was developed to detect Acetilactobacillus jinshanensis, a dominant functional microorganism in Jiang-flavour Baijiu production. This assay achieved a remarkable sensitivity of 1 copy/μL, surpassing traditional qPCR methods. When applied to actual fermentation samples, the biosensor revealed a direct correlation between A. jinshanensis concentration and base liquor quality, with excellent-grade workshops showing average fluorescence values of 4838 compared to 2145 in ordinary workshops [83]. This demonstrates how automated CRISPR detection can predict final product quality during early fermentation stages.
Pathogen Detection in Fermented Foods
Fermented foods remain vulnerable to pathogen contamination, necessitating robust monitoring solutions. The hMC-CRISPR platform was specifically designed to detect pathogenic Listeria species (L. monocytogenes, L. innocua, and L. ivanovii) with attomolar sensitivity. This system employs Cas13a with specifically designed crRNAs targeting unique genetic markers of each species, enabling multiplexed detection without cross-reactivity [84]. The platform successfully identified Listeria in 24 natural samples with high accuracy, demonstrating its practical utility for food safety monitoring in fermentation facilities. The closed-system design prevents amplicon contamination, a critical advantage for routine quality control testing [84].
Process Control and Optimization
Integrating CRISPR biosensors with Internet of Things (IoT) devices and machine learning algorithms creates intelligent fermentation monitoring systems. These systems can track microbial population dynamics in real-time, enabling proactive intervention when deviations from optimal profiles occur. Smart fermentation technologies help bridge the gap between traditional artisanal methods and modern industrial production by providing data-driven insights while preserving microbial biodiversity and cultural heritage [26]. The implementation of affordable IoT devices and open-source platforms makes this technology increasingly accessible to small-scale producers traditionally dominating fermented food production [26].
Detailed Protocols
Protocol 1: TRACE One-Pot Assay for Microbial Detection
This protocol adapts the TRACE assay for detecting specific microbes in food fermentation samples [81].
Reagents and Equipment:
- Cas12b protein (100 µM)
- crRNA specific to target microbe (200 µM)
- ssRNA blocker (200 µM, 15°C lower Tm than reaction temperature)
- RPA basic kit (TwistAmp)
- Fluorescent reporter (e.g., ssDNA with 6-FAM/BHQ-1)
- Thermal cycler or heat blocks (37°C and 60°C)
- Food fermentation samples
Procedure:
- Reaction Mixture Preparation:
- Prepare RPA reaction mix according to manufacturer's instructions
- Add Cas12b:crRNA complex at 400 nM final concentration (pre-formed by incubating 15 min at room temperature)
- Include ssRNA blocker at 1:4 ratio (gRNA:blocker)
- Add fluorescent reporter (400 nM final concentration)
- Include sample DNA (1-10 µL depending on concentration)
Thermal Cycling:
- Incubate at 37°C for 10 minutes (RPA amplification phase)
- Transfer to 60°C for 30 minutes (CRISPR detection phase)
- Monitor fluorescence in real-time or measure endpoint fluorescence
Data Interpretation:
- Compare fluorescence to standard curve from known concentrations
- Include positive and negative controls in each run
- Threshold: Signal > 3x standard deviation of negative control
Optimization Notes:
- Test ssRNA blockers with varying lengths to achieve optimal inhibition during amplification phase
- For different microbial targets, redesign crRNA while maintaining optimal secondary structure
- Adjust RPA incubation time based on target abundance (extend to 15-20 min for very low targets)
Protocol 2: Automated CRISPR Workflow on Modular Platform
This protocol outlines automated processing for high-throughput microbial detection in fermentation monitoring [85].
Reagents and Equipment:
- Automated platform (e.g., StemCellFactory with COPE software)
- 96-well nucleofector plates
- P3 nucleofection buffer
- Cas12a/gRNA complex (pre-formed)
- RPA reagents
- Sample plates containing fermentation extracts
Procedure:
- System Initialization:
- Calibrate robotic arm and liquid handling systems
- Load consumables (tip boxes, nucleofector plates, reagent reservoirs)
- Prime liquid handling lines with appropriate buffers
Reaction Setup:
- Program automated method to distribute 19.5 µL P3 buffer to each well
- Add 4 µL pre-formed Cas12a/gRNA complex to each well
- Transfer 3×10^5 cells (or microbial equivalents) per condition in 1 µL volume
- Mix by automated pipetting (5 cycles of 30 µL aspiration/dispense)
Nucleofection and Detection:
- Transfer nucleofector plate to 4D-Nucleofector system
- Execute program CM150 for electroporation
- Immediately add 100 µL recovery medium with RPA reagents
- Transfer to incubation station at 37°C for 30-40 min
- Measure fluorescence using integrated plate reader
Data Analysis and Reporting:
- Automated data capture and analysis through COPE software
- Generation of sample reports with concentration estimates
- Flagging of abnormal results for manual review
Automation Notes:
- Implement two-stage error handling for robust operation
- Use deep learning algorithms for automated quality control of signals
- Maintain sample tracking through barcode identification systems
The integration of one-pot assay designs and automation platforms has significantly advanced CRISPR-based biosensing for food fermentation research. These technologies provide the sensitivity, specificity, and throughput required to monitor complex microbial communities in dynamic fermentation environments. The TRACE assay's thermal regulation, microfluidic systems' multiplexing capabilities, and automated platforms' standardization represent significant milestones toward making robust microbial detection more accessible and reliable. As these technologies continue to evolve, they will play an increasingly vital role in ensuring fermented product quality, safety, and consistency while preserving the traditional practices that give these foods their unique characteristics. Future developments will likely focus on reducing costs, enhancing multiplexing capabilities, and improving user-friendliness for broader adoption across the food fermentation industry.
Benchmarking Performance: Validation Against Gold Standards and Future Potential
In the field of food fermentation research, the accurate and timely monitoring of microbial communities is paramount for ensuring product quality, safety, and consistency. Traditional detection methods, primarily culture-based techniques and polymerase chain reaction (PCR) or quantitative PCR (qPCR), have long been the standard. However, the emergence of CRISPR-based biosensors presents a transformative approach, offering significant advancements in speed, sensitivity, and specificity [34] [5]. This application note provides a structured comparison of these methodologies, details an optimized protocol for CRISPR-based detection, and visualizes the essential components for implementation in a research setting.
The core advantage of CRISPR diagnostics lies in its programmable nature and unique enzymatic activity. Systems utilizing Cas12 and Cas13 effectors demonstrate exceptional specificity through crRNA-guided target recognition, coupled with high sensitivity enabled by trans-cleavage activity that amplifies detection signals [34] [6]. This combination allows for the precise identification of microbial targets, such as starter cultures, spoilage organisms, or pathogens, within complex food matrices like fermenting products.
Comparative Performance Data
The following tables summarize key performance metrics for CRISPR-based detection compared to traditional methods, based on recent meta-analyses and validation studies.
Table 1: Overall Diagnostic Accuracy of CRISPR-based Methods for Pathogen Detection
| Method | Pooled Sensitivity (%) | Pooled Specificity (%) | Diagnostic Odds Ratio (DOR) | Primary Application in Review |
|---|---|---|---|---|
| CRISPR-based Detection | 99 (95% CI: 97-100) | 100 (95% CI: 99-100) | 664.25 | MRSA detection in clinical samples [86] |
| Culture-Based Methods | >99* | >99* | N/A | Considered gold standard but slow [5] |
| PCR/qPCR | High (varies by assay) | High (varies by assay) | N/A | Widely used molecular standard [34] |
*Culture-based methods are considered a reference for specificity but can miss viable but non-culturable (VBNC) organisms [18].
Table 2: Practical Workflow and Sensitivity Comparison
| Parameter | Culture-Based Methods | PCR/qPCR | CRISPR-Based Biosensors |
|---|---|---|---|
| Typical Time-to-Result | 2-5 days [86] | 2-4 hours [86] | ~60 minutes (IQR: 41-99 min) [86] |
| Limit of Detection (LOD) | Varies (10-100 CFU) [5] | High (attomolar levels) [34] | Extremely High (attomolar, e.g., 20 aM) [87] |
| Equipment Needs | Incubators, biosafety cabinets | Thermal cyclers, qPCR machines | Minimal; potential for portable readers [5] |
| Ease of Use | Labor-intensive, requires expertise | Requires trained personnel | Simpler; amenable to point-of-care [88] |
| Multiplexing Potential | Low | Moderate | High (e.g., 9-plex platforms) [89] |
A systematic review and meta-analysis focused on Methicillin-Resistant Staphylococcus aureus (MRSA) detection demonstrated the exceptional accuracy of CRISPR-based methods, with a pooled sensitivity of 99% and specificity of 100%, significantly outperforming conventional methods in speed with a median detection time of 60 minutes [86]. Furthermore, CRISPR biosensors can achieve astonishing sensitivity without target amplification, such as an impedimetric biosensor detecting Staphylococcus aureus DNA at concentrations as low as 20 attomolar (aM) [87].
Detailed Experimental Protocol for RPA-CRISPR/Cas12a-based Detection
This protocol describes a rapid, sensitive method for detecting specific bacterial DNA in a food fermentation sample, leveraging recombinase polymerase amplification (RPA) and CRISPR/Cas12a activation, adapted from established clinical diagnostics [88] [90].
Safety and Pre-Planning
- Biosafety: Adhere to BSL-2 practices for handling potentially pathogenic microorganisms.
- Contamination Control: Use dedicated rooms/hoods for pre- and post-amplification steps, employ UV decontamination, and use filter tips to prevent aerosol contamination.
Sample Preparation and DNA Extraction
- Sample Collection: Aseptically collect 1 mL of fermenting broth.
- Cell Lysis: Transfer 100 µL of broth to a tube containing 100 µL of nucleic acid lysis buffer (e.g., from Shanghai Kanglang Biotechnology). Vortex vigorously and incubate at room temperature for 5 minutes [88].
- Crude Extraction: Centrifuge the lysate at 12,000 × g for 2 minutes. The supernatant containing the crude DNA extract can be used directly as the template for RPA. For complex matrices, consider using commercial DNA extraction kits for higher purity.
Primer and crRNA Design
- Target Selection: Identify a highly conserved, unique gene sequence from the target microbe (e.g., 16S rRNA, gyrB, or a species-specific virulence gene).
- RPA Primer Design: Design primers approximately 30-35 nucleotides long with a melting temperature (Tm) of around 60-65°C, targeting a 100-300 bp amplicon. Use software (e.g., Primer Premier) and the TwistAmp assay design manual. Verify specificity via NCBI-BLAST.
- crRNA Design: Design a ~20-25 nt spacer sequence within the RPA amplicon, ensuring it is adjacent to a suitable Protospacer Adjacent Motif (PAM) for Cas12a (e.g., 5'-TTTV-3' for LbCas12a). Use online tools like Benchling for design and ClustalW for conservancy analysis [88].
Recombinase Polymerase Amplification (RPA)
- Prepare RPA Reaction: On ice, assemble a 50 µL reaction using a commercial kit (e.g., TwistAmp Basic):
- 29.5 µL of rehydration buffer
- Forward and Reverse Primers (10 µM each)
- 5 µL of the crude DNA extract (template)
- Nuclease-free water to 50 µL
- Initiate Amplification: Add a magnesium acetate pellet (provided in the kit) to the tube, quickly spin down, and immediately place in a dry block heater at 39°C for 15-20 minutes [88].
CRISPR/Cas12a Detection and Signal Readout
- Prepare CRISPR Reaction Mix: For each reaction, combine:
- 4 µL of EnGen Lba Cas12a (1 µM final)
- 2 µL of crRNA (1 µM final)
- 2 µL of NEBuffer 2.1 (1× final)
- 1 µL of ssDNA Fluorescent Reporter (e.g., FAM-TTATT-BHQ1, 500 nM final)
- 6 µL of nuclease-free water
- Activate Detection: Add 5 µL of the RPA amplicon directly into the 15 µL CRISPR reaction mix. Mix gently and incubate at 37°C for 10-15 minutes.
- Visualize Results:
- Fluorescence: Transfer the reaction to a tube or plate and visualize under a portable blue light transilluminator (470 nm). A bright green fluorescent signal indicates a positive result [88].
- Colorimetric (Alternative): For a colorimetric readout, a more complex probe (e.g., MNPs-ssDNA-HRP) can be used. The activated Cas12a cleaves the probe, releasing HRP, which then catalyzes a TMB substrate, causing a color change from colorless to blue [90].
Signaling Pathways and Workflows
The following diagram illustrates the core molecular mechanism of the Cas12a-based detection system described in this protocol.
Figure 1: Cas12a Detection Mechanism. The Cas12a protein, guided by a crRNA, binds to the target double-stranded DNA (dsDNA) to form a ternary complex. This binding activates the protein's non-specific trans-cleavage activity, which indiscriminately degrades single-stranded DNA (ssDNA) reporters. Cleavage of the reporter separates a fluorophore (F) from a quencher (Q), generating a detectable fluorescent signal [34] [6] [90].
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Reagents and Materials for RPA-CRISPR/Cas12a Assay
| Item | Function/Description | Example Supplier / Catalog |
|---|---|---|
| Lba Cas12a Protein | CRISPR effector enzyme for target-specific binding and trans-cleavage. | New England Biolabs (EnGen Lba Cas12a) [88] [90] |
| crRNA | Custom RNA guide that directs Cas12a to the specific target DNA sequence. | Synthesized by companies like Sangon Biotech [90] |
| RPA Kit | Isothermal amplification kit for rapid, low-temperature nucleic acid amplification. | TwistDx (TwistAmp Basic) [88] |
| Fluorescent ssDNA Reporter | ssDNA oligonucleotide with a fluorophore and quencher; cleavage produces signal. | FAM-TTATT-BHQ1 (Sangon Biotech) [88] |
| Nucleic Acid Lysis Buffer | Rapidly lyses microbial cells to release nucleic acids for direct amplification. | Shanghai Kanglang Biotechnology [88] |
| Portable Blue Light Transilluminator | Enables visual, on-site fluorescence readout without complex instrumentation. | Various suppliers (470 nm excitation) [88] |
| Terminal Deoxynucleotidyl Transferase (TdT) | For multimodal detection: Enzymatically synthesizes poly-T strands for copper nanocluster formation. | Takara Biotech [90] |
| Horseradish Peroxidase (HRP) | For colorimetric detection: Enzyme that catalyzes TMB for color change. | Beyotime Biotechnology (SA/HRP) [90] |
The integration of Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR) technology into biosensing platforms has revolutionized diagnostic methodologies, offering a powerful tool for microbial detection in food fermentation research. For scientists and drug development professionals, the critical metrics of Limit of Detection (LoD), turnaround time, and cost-effectiveness serve as pivotal parameters for evaluating assay performance and practical applicability. CRISPR-based biosensors leverage the programmability of CRISPR RNA (crRNA) and the enzymatic activity of Cas proteins to achieve unprecedented specificity and sensitivity in detecting nucleic acid targets. These systems have demonstrated remarkable capabilities, with certain configurations achieving attomolar (aM) sensitivity and delivering results in under 30 minutes, making them particularly suitable for quality control and contamination monitoring in fermentation processes [68] [34]. This application note provides a comprehensive analysis of these key performance metrics across major CRISPR platforms, along with detailed experimental protocols tailored for food fermentation research applications.
Performance Metrics of Major CRISPR-Cas Systems
Comparative Analysis of Cas Effectors
Table 1: Performance Characteristics of CRISPR-Cas Effectors in Diagnostic Applications
| Cas Protein | Target Nucleic Acid | Trans-cleavage Activity | Typical LoD | Key Applications | PAM Requirement |
|---|---|---|---|---|---|
| Cas9 | DNA/RNA | None (cis-cleavage only) | Medium | Laboratory research, SNP detection | NGG |
| Cas12a (Cpf1) | DNA | ssDNA | High (aM-fM) | DNA pathogen detection, viral identification | TTTV |
| Cas13a | RNA | ssRNA | High (aM-fM) | RNA virus detection, miRNA profiling | None |
| Cas14 (Cas12f) | ssDNA | ssDNA | Very High | SNP detection, short ssDNA targets | None |
CRISPR-Cas systems are categorized into two principal classes based on their effector complex structure. Class 1 (types I, III, and IV) utilizes multi-subunit crRNA-effector complexes, while Class 2 (types II, V, and VI) employs single protein effectors, which have become the cornerstone of modern CRISPR diagnostics due to their simpler architecture [68] [91]. The fundamental mechanism underpinning CRISPR diagnostics involves the programmable recognition of target nucleic acids through complementary crRNA, leading to Cas enzyme activation. For certain effectors like Cas12 and Cas13, this activation triggers robust trans-cleavage activity – a nonspecific degradation of surrounding reporter molecules that generates amplified, detectable signals [34] [92].
The selection of an appropriate Cas effector is guided by the nature of the target analyte. Cas12a effectors, which target DNA and exhibit collateral cleavage of single-stranded DNA (ssDNA) reporters, are ideal for detecting DNA-based microbial contaminants in fermentation substrates [92]. Conversely, Cas13a systems, which target RNA and demonstrate collateral activity against single-stranded RNA (ssRNA), are exceptionally suited for monitoring RNA viruses or profiling microbial community activity through mRNA expression analysis [39] [92]. The Cas14 protein, with its preference for single-stranded DNA targets and lack of protospacer adjacent motif (PAM) sequence requirements, offers unique advantages for detecting small genetic targets or single-nucleotide polymorphisms (SNPs) without amplification [68] [93].
Key Performance Metrics Analysis
Table 2: Quantitative Performance Metrics of CRISPR Detection Platforms
| CRISPR Platform | Combined Technology | Reported LoD | Turnaround Time | Sample Targets | Key Advantages |
|---|---|---|---|---|---|
| Cas12a-DETECTR | RPA pre-amplification | 1 copy/µL | 30-60 minutes | Mpox DNA, Bacteria | High DNA sensitivity, suitable for POC |
| Cas13-SHERLOCK | RPA pre-amplification | aM range | <60 minutes | SARS-CoV-2, miRNAs | Superior RNA detection, high specificity |
| Cas12b-HOLMES | LAMP pre-amplification | aM range | ~1 hour | Viruses, Bacteria | Stable at higher temperatures |
| Amplification-free Cas13a | Direct detection | 470 aM | <30 minutes | SARS-CoV-2, HIV-1 | Simplified workflow, minimal equipment |
| One-pot RPA-CRISPR | Integrated RPA/CRISPR | fM range | 20-40 minutes | Various pathogens | Reduced contamination, faster results |
The Limit of Detection (LoD) for CRISPR-based biosensors varies significantly based on the Cas effector employed and whether nucleic acid amplification is incorporated. Systems coupled with pre-amplification techniques like Recombinase Polymerase Amplification (RPA) or Loop-Mediated Isothermal Amplification (LAMP) routinely achieve exceptional sensitivity, detecting targets at attomolar (10⁻¹⁸ M) concentrations or even single copies of microbial genomes [68] [91]. For instance, the integration of RPA with CRISPR-Cas12a has demonstrated detection of Mpox DNA at sensitivities as low as 1 copy per microliter [68]. Similarly, amplification-free CRISPR strategies have achieved remarkable sensitivity, with one Cas13a platform reporting an LoD of 470 aM for SARS-CoV-2 detection within 30 minutes, highlighting the potential for rapid testing without compromising sensitivity [68].
Turnaround time represents another critical metric, particularly for fermentation monitoring applications requiring rapid intervention. Traditional PCR-based methods typically require 2-4 hours due to thermal cycling requirements, whereas CRISPR-based assays leveraging isothermal amplification can deliver results in under 60 minutes [34] [91]. The emergence of one-pot detection systems, which integrate amplification and CRISPR detection in a single closed tube, has further reduced processing times to 20-40 minutes while simultaneously minimizing contamination risks by eliminating reagent transfer steps [91].
The cost-effectiveness of CRISPR diagnostics stems from multiple factors: minimal reagent requirements, compatibility with inexpensive portable readers, and reduced dependency on sophisticated laboratory infrastructure. The operational simplicity of these assays aligns with the World Health Organization's ASSURED criteria (Affordable, Sensitive, Specific, User-friendly, Rapid and robust, Equipment-free, and Deliverable to end-users), making them particularly suitable for resource-limited settings [91] [94]. The development of lyophilized reagents and lateral flow readouts has further enhanced deployment capability while maintaining low per-test costs [34] [94].
Experimental Protocols for Food Fermentation Applications
One-Pot RPA-CRISPR/Cas12a Protocol for Microbial Contamination Detection
This protocol describes an integrated approach for detecting bacterial contaminants in fermentation samples using a combined RPA-CRISPR/Cas12a system in a single tube, significantly reducing processing time and contamination risk.
Workflow Diagram: One-Pot RPA-CRISPR Detection Protocol
Reagents and Materials
- Cas12a enzyme (commercial sources such as New England Biolabs or IDT)
- crRNA designed against target microbial DNA sequence (see Section 3.1.2 for design guidelines)
- RPA primers targeting conserved microbial genes (e.g., 16S rRNA, specific virulence factors)
- ssDNA reporter molecule (e.g., FAM-TTATT-BHQ or FAM-TTATT-IBFQ for fluorescence quenching)
- RPA basic reaction kit (TwistDx Ltd. or comparable supplier)
- Magnesium acetate (280mM for RPA reaction initiation)
- Nuclease-free water
- Fermentation sample (1-5 mL liquid culture or 0.1-0.5 g solid substrate)
crRNA Design Guidelines
- Identify a unique genomic region in the target microorganism with no significant homology to non-target species potentially present in fermentation samples
- Ensure the target sequence contains a compatible PAM site (TTTV for Cas12a, where V is A, G, or C) adjacent to the target region
- Design crRNA spacer sequences of approximately 20-24 nucleotides with minimal secondary structure
- Verify specificity using BLAST analysis against relevant genomic databases
- For single-nucleotide specificity, position the discriminatory base within the seed region (positions 3-10 from PAM) of the spacer sequence [93]
Step-by-Step Procedure
Sample Preparation
- For liquid fermentation samples: Centrifuge 1 mL at 8,000 × g for 5 minutes, resuspend pellet in 100 µL nuclease-free water
- For solid fermentation substrates: Homogenize 0.1 g sample in 1 mL PBS, centrifuge at 8,000 × g for 5 minutes, collect supernatant
Nucleic Acid Extraction
- Use commercial DNA extraction kits following manufacturer's instructions
- Elute DNA in 30-50 µL elution buffer
- Quantify DNA concentration using spectrophotometry (optional for qualitative detection)
One-Pot RPA-CRISPR Reaction Setup
- Prepare master mix in a single tube:
- 29.5 µL rehydration buffer (from RPA kit)
- 2.4 µL forward primer (10 µM)
- 2.4 µL reverse primer (10 µM)
- 1 µL crRNA (10 µM)
- 1 µL Cas12a enzyme (1 µM)
- 1 µL ssDNA reporter (10 µM)
- 2.7 µL nuclease-free water
- 5 µL template DNA
- Transfer 45 µL of the mixture to a 0.2 mL PCR tube
- Add 5 µL magnesium acetate (280 mM) to the tube lid, then briefly centrifuge to initiate reaction
- Prepare master mix in a single tube:
Incubation and Detection
- Incubate reaction at 37-42°C for 20-40 minutes
- Monitor fluorescence in real-time using a portable fluorometer or endpoint measurement using a lateral flow strip
- For lateral flow detection: Apply 5 µL reaction mixture to the sample pad, insert into running buffer, wait 5 minutes for visual development
Data Interpretation
- Positive result: Significant fluorescence increase above background threshold or visible test line on lateral flow strip
- Negative result: No fluorescence increase or absence of test line (control line should be visible for valid test)
- Quantitative analysis: Generate standard curve using known concentrations of target DNA for absolute quantification
Amplification-Free CRISPR/Cas13a Protocol for RNA Biomarker Detection
This protocol enables direct detection of RNA biomarkers without amplification, ideal for monitoring microbial metabolic activity or RNA viral contaminants in fermentation processes where speed is critical.
Reagents and Materials
- Cas13a enzyme (commercial sources)
- crRNA designed against target RNA sequence
- ssRNA reporter molecule (e.g., FAM-UUUU-BHQ or comparable quenched fluorescent reporter)
- RNA extraction kit with DNase treatment capability
- RNase inhibitors
- Nuclease-free water and tubes
Step-by-Step Procedure
RNA Extraction from Fermentation Samples
- Process samples as in Section 3.1.3, step 1
- Extract RNA using commercial kits with DNase treatment to eliminate DNA contamination
- Elute RNA in 20-30 µL nuclease-free water
- Maintain samples on ice to preserve RNA integrity
CRISPR/Cas13a Reaction Setup
- Prepare reaction mix:
- 2 µL Cas13a (1 µM)
- 2 µL crRNA (10 µM)
- 1 µL ssRNA reporter (10 µM)
- 2 µL RNase inhibitor
- 8 µL reaction buffer (provided with Cas13a)
- 5 µL RNA template
- 30 µL total volume with nuclease-free water
- Mix gently by pipetting, avoid vortexing
- Prepare reaction mix:
Incubation and Detection
- Incubate at 37°C for 15-30 minutes
- Measure fluorescence at regular intervals (2-5 minutes) using a portable fluorometer
- Compare endpoint fluorescence to negative controls
Data Analysis
- Calculate ΔRFU (Relative Fluorescence Units) by subtracting background fluorescence
- Establish positive threshold at 3× standard deviation above negative control mean
- For quantitative applications, generate standard curve with in vitro transcribed RNA targets
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Research Reagent Solutions for CRISPR-Based Microbial Detection
| Reagent Category | Specific Examples | Function in Assay | Considerations for Fermentation Research |
|---|---|---|---|
| Cas Enzymes | Cas12a, Cas13a, Cas14 | Sequence-specific target recognition and trans-cleavage activity | Select based on target (DNA vs. RNA); Cas12a preferred for DNA targets in bacterial detection |
| Guide RNAs | Custom crRNAs, sgRNAs | Programmable recognition element guiding Cas to target | Design against unique microbial genomic regions; ensure compatibility with fermentation matrix |
| Reporter Molecules | FAM-TTATT-BHQ (ssDNA), FAM-UUUU-BHQ (ssRNA) | Signal generation upon trans-cleavage | Quencher selection affects background; optimize concentration to maximize signal-to-noise |
| Amplification Reagents | RPA, LAMP kits | Pre-amplification to enhance sensitivity | RPA offers faster results; LAMP provides robust amplification; choose based on temperature requirements |
| Sample Preparation Kits | DNA/RNA extraction kits | Nucleic acid isolation from complex matrices | Select kits effective for fermentation samples; include DNase treatment for RNA targets |
| Readout Systems | Lateral flow strips, portable fluorometers | Result visualization and quantification | Lateral flow for field use; fluorometers for quantification; consider integration with fermentation monitoring systems |
CRISPR-based biosensors represent a transformative technology for microbial detection in food fermentation research, offering an exceptional combination of sensitivity, speed, and practical implementation. The quantitative metrics presented in this analysis demonstrate that these systems can achieve detection limits rivaling traditional PCR while significantly reducing turnaround times from hours to minutes. The intrinsic cost-effectiveness and portability of CRISPR-based platforms further enhance their suitability for routine fermentation monitoring, quality control, and contamination detection. As CRISPR diagnostics continue to evolve through innovations such as one-pot reactions, amplification-free strategies, and multiplex detection capabilities, their integration into fermentation research workflows promises to accelerate discovery and improve process control. The experimental protocols provided herein offer researchers comprehensive guidelines for implementing these powerful tools in their microbial detection applications.
The ASSURED criteria, established by the World Health Organization, provide a benchmark for ideal diagnostic tools in resource-limited settings, defining them as Affordable, Sensitive, Specific, User-friendly, Rapid and robust, Equipment-free, and Deliverable to end-users [95] [96]. For researchers in food fermentation, applying these criteria to emerging detection technologies ensures that developed methods are practical for real-time monitoring within production facilities. CRISPR-based biosensors represent a transformative advancement in this domain, leveraging the programmable nucleic acid recognition capabilities of CRISPR-Cas systems to detect microbial contaminants and monitor fermentation processes with exceptional precision [5] [95].
The integration of CRISPR diagnostics into food fermentation research addresses critical gaps in traditional microbial detection methods. Culture-based techniques, while reliable, require days to yield results and are unsuitable for real-time decision-making [5]. Molecular methods like PCR, though faster, often necessitate sophisticated laboratory infrastructure and trained personnel, limiting their application at the point-of-care [97] [96]. CRISPR-based systems bridge this gap by combining laboratory-grade accuracy with the potential for field-deployment, making them particularly valuable for ensuring the microbial safety and quality of fermented products [5] [18].
Quantitative ASSURED Performance of CRISPR-Based Biosensors
The performance of CRISPR-based biosensors against the ASSURED framework reveals their strong suitability for food fermentation monitoring applications. The table below summarizes key quantitative metrics reported in recent studies.
Table 1: Performance Metrics of CRISPR-Based Biosensors Against ASSURED Criteria
| ASSURED Criteria | Performance Metrics | Typical Values/Characteristics | References |
|---|---|---|---|
| Affordable | Estimated cost per test | Potentially < USD 1 in some formats | [95] |
| Sensitive | Limit of Detection (LoD) | As low as 0.82 amol (synthetic DNA); 1–50 CFU/mL for bacteria | [5] [98] |
| Specific | Single-nucleotide discrimination | Capable of distinguishing highly homologous sequences | [99] [98] |
| User-friendly | Total assay steps | Minimal processing; results interpretable via color change or lateral flow strip | [95] [98] |
| Rapid & Robust | Total turnaround time | 30 minutes to 4 hours (significantly faster than culture methods) | [100] [101] |
| Equipment-free | Need for specialized instruments | Can be integrated with portable readers or visual readouts; minimal equipment | [5] [95] |
| Deliverable | Stability and portability | Stable reagents; lyophilized formats possible; deployable in field settings | [95] |
The data demonstrates that CRISPR platforms meet multiple ASSURED criteria simultaneously. Their high sensitivity and specificity are comparable to laboratory-based PCR methods, while their rapid turnaround time—orders of magnitude faster than traditional culture methods that can take 5-7 days—enables near real-time monitoring of microbial populations in fermenting products [5] [96]. This speed is critical for implementing timely corrective actions during fermentation processes.
Experimental Protocol for On-Site Microbial Detection in Food Fermentation
This protocol details a standard workflow for detecting a specific bacterial target (e.g., Listeria monocytogenes or a spoilage organism) in a fermented food matrix using a CRISPR-Cas12a system, integrating pre-amplification and lateral flow readout for point-of-care applicability.
Research Reagent Solutions
Table 2: Essential Reagents and Materials for CRISPR-Based Detection
| Item | Function/Description | Application Note |
|---|---|---|
| LbaCas12a or LbCas12a Enzyme | Type V CRISPR effector; provides target-specific dsDNA recognition and collateral ssDNA cleavage. | The most commonly used Cas enzyme in bacterial detection (64% of studies) [100] [101]. |
| Target-specific crRNA | Guide RNA complementary to the target DNA sequence (e.g., hly gene of L. monocytogenes). | Dictates the specificity of the assay; must be designed to target a unique genomic region. |
| ssDNA Fluorescent Reporter (e.g., FQ-ssDNA) | Reporter probe (e.g., 6-FAM-TTATTATT-BHQ1); cleavage generates fluorescent signal. | For real-time fluorescence detection using a portable reader. |
| Lateral Flow Strip Reporter (e.g., Biotin-ssDNA-FAM) | Reporter probe for lateral flow readout; cleavage prevents test line capture. | Enables visual, equipment-free result interpretation. |
| Recombinase Polymerase Amplification (RPA) Kit | Isothermal nucleic acid amplification; operates at 37–42°C. | The most common isothermal method used with CRISPR detection (66% of studies use isothermal amplification) [100] [101]. |
| Nucleic Acid Extraction Kit | Prepares template DNA from the complex food matrix. | Critical step; efficiency can be impacted by food components [5]. |
| Portable Fluorescence Reader or Lateral Flow Strips | Signal detection device. | Meets the "Equipment-free" or "Deliverable" criterion for low-resource settings. |
Step-by-Step Workflow
Step 1: Sample Collection and Nucleic Acid Extraction. Collect a representative sample (e.g., 1 mL of fermenting broth or 1 g of solid product). Extract genomic DNA using a commercial kit suitable for the specific food matrix. The extraction step is crucial, as inhibitors present in complex food matrices (fats, proteins, carbohydrates) can reduce the efficiency of both amplification and CRISPR detection, potentially leading to false-negative results [5]. The final elution volume should be 30-50 µL.
Step 2: Target Pre-amplification via RPA. Prepare a 50 µL RPA reaction mix according to the manufacturer's instructions. It typically contains:
- 29.4 µL of rehydration buffer
- 2.4 µL of forward primer (10 µM)
- 2.4 µL of reverse primer (10 µM)
- 12.5 µL of the extracted DNA template
- 2.5 µL of magnesium acetate (280 mM) Add the primers specific to the target gene. Incubate the reaction at 39°C for 15–20 minutes. Isothermal amplification methods like RPA are preferred over PCR as they do not require thermal cycling, aligning with the equipment-free criterion [5] [95].
Step 3: CRISPR-Cas12a Detection. Prepare a 20 µL CRISPR reaction mixture:
- 2 µL of 10x Cas12a reaction buffer
- 1 µL of LbaCas12a enzyme (10 µM)
- 1.5 µL of crRNA (10 µM)
- 1 µL of ssDNA reporter (10 µM, e.g., FQ-ssDNA for fluorescence or Biotin-ssDNA-FAM for lateral flow)
- 14.5 µL of nuclease-free water Incubate this mixture at 37°C for 5 minutes. Then, add 2 µL of the RPA amplicon from Step 2, mix gently, and incubate at 37°C for an additional 10–15 minutes. The activated Cas12a will collaterally cleave the reporter probes, generating a detectable signal.
Step 4: Result Readout and Interpretation.
- Fluorescence Readout: Transfer the reaction tube to a portable fluorescence reader. A significant increase in fluorescence intensity over a negative control indicates a positive detection.
- Lateral Flow Readout: For a visual result, apply the reaction mixture to a lateral flow strip. In a positive test, the cleaved reporter cannot be captured at the test line, leading to only the control line appearing. The appearance of both control and test lines indicates a negative result. The absence of the control line indicates an invalid test.
Assessment Against ASSURED Framework
Affordability and Deliverability
The core reagents for CRISPR-based detection (Cas enzyme, crRNA, reporters) are becoming increasingly affordable due to streamlined production. The potential for lyophilizing reaction components into stable, room-temperature pellets enhances deliverability to remote or resource-limited fermentation facilities, eliminating the cold chain requirement [95]. The integration with low-cost RPA and visual lateral flow readouts keeps the overall cost per test competitive, potentially under USD 1 for high-volume applications [95].
Speed and Equipment Needs
The total assay time of under 60 minutes fulfills the "Rapid" criterion, providing a decisive advantage over culture methods (5-7 days) and even some PCR protocols [100] [96]. The protocol's reliance on a single, low-temperature incubation block (or even body heat for RPA) and the option for visual readout significantly reduce equipment dependence, meeting the "Equipment-free" and "Deliverable" goals [5] [95]. This makes the technology suitable for on-site use in fermentation plants without central laboratories.
Sensitivity, Specificity, and User-friendliness
CRISPR-biosensors achieve high sensitivity (single-digit CFU/mL levels) primarily through the combination of pre-amplification and the CRISPR system's catalytic signal amplification [5] [98]. Their specificity is exceptional, capable of distinguishing between closely related microbial strains, which is vital for identifying specific starter cultures or contaminants within a complex fermenting microbiome [99]. The move towards all-in-one-tube reactions and simple visual readouts (lateral flow strips) makes the assays user-friendly, requiring minimal technical expertise [95] [98].
CRISPR-based biosensors present a powerful diagnostic platform that aligns strongly with the ASSURED criteria, making them exceptionally suitable for integration into food fermentation research and monitoring. Their speed, sensitivity, and specificity address the critical need for real-time microbial analysis during fermentation processes. Furthermore, their evolving affordability, minimal equipment needs, and user-friendly formats pave the way for their deployment directly in production facilities, ultimately contributing to enhanced food safety, optimized fermentation control, and higher quality fermented products.
CRISPR-based biosensors represent a transformative advancement in food fermentation research, merging the precise targeting capabilities of CRISPR-Cas systems with sensitive signal transduction mechanisms. These tools address critical limitations of conventional microbiological methods, which are often time-consuming and labor-intensive, by enabling rapid, specific, and on-site detection of microbial populations and contaminants [18] [50]. This analysis examines the deployment of these biosensors across three key fermentation sectors—dairy, meat, and plant-based—highlighting their operational principles, validated performance, and detailed protocols to guide research and development professionals.
Technical Foundation of CRISPR Biosensors
Core CRISPR-Cas Protein Mechanisms
The functionality of CRISPR biosensors in food diagnostics hinges on the distinct biochemical activities of Class II Cas proteins, which can be programmed to recognize specific nucleic acid sequences.
- Cas9 operates through a cis-cleavage activity, creating double-strand breaks in target double-stranded DNA (dsDNA) upon recognition of a protospacer adjacent motif (PAM) sequence. While useful, its lack of trans-cleavage activity limits its signal amplification potential in biosensing [102] [33].
- Cas12 (including subtypes Cas12a) and Cas14 target DNA. Upon binding and cleaving its target dsDNA (a process guided by a CRISPR RNA, crRNA), it exhibits nonspecific trans-cleavage activity, indiscriminately degrading single-stranded DNA (ssDNA) reporters in the solution. This collateral cleavage is highly effective for signal amplification in biosensors [79] [50] [33].
- Cas13 targets RNA and, similar to Cas12, possesses trans-cleavage activity. Once activated by its target RNA, it cleaves surrounding non-specific RNA molecules, providing a powerful mechanism for RNA detection and signal amplification [79] [33].
The trans-cleavage activity of Cas12 and Cas13 is the cornerstone of most current CRISPR-based detection platforms, as it allows a single target recognition event to trigger numerous reporter cleavage events, thereby significantly boosting detection sensitivity [50].
Biosensor Signal Transduction and Readout Modalities
The collateral cleavage of reporter molecules by activated Cas proteins can be converted into a detectable signal through various transduction methods, listed in Table 1.
Table 1: Common Signal Transduction Methods in CRISPR Biosensors
| Readout Method | Signal Reported | Reporter Molecule Example | Advantages | Typical Limit of Detection |
|---|---|---|---|---|
| Fluorescence | Fluorescence intensity | FAM-labeled ssDNA quenched by BHQ1 [102] | High sensitivity, suitability for quantification | aM (10⁻¹⁸ M) to fM (10⁻¹⁵ M) [102] |
| Electrochemical (EC) | Change in current, potential, or impedance | Methylene Blue (MB)-labeled ssDNA [50] | High sensitivity, portability, miniaturization potential | ~0.634 pM (amplification-free) [50] |
| Colorimetric | Visible color change | Gold Nanoparticles (AuNPs) or colorimetric substrates [33] | Simplicity, equipment-free, naked-eye readout | Varies; can achieve visual aM detection [33] |
The following diagram illustrates the core mechanism of Cas12a/Cas13a-based fluorescent biosensing.
Application Notes: Sector-Specific Deployments
Dairy Fermentation
In dairy production, CRISPR biosensors are pivotal for ensuring starter culture vitality and preventing pathogen contamination.
- Pathogen Detection: A primary application is the rapid detection of Listeria spp. and E. coli.
- Starter Culture Monitoring: Biosensors enable real-time tracking of essential lactic acid bacteria (LAB) like Lactobacillus plantarum during fermentation. Isothermal microcalorimetry-based detection has demonstrated quantification within a range of 4.7–18.6 hours, ensuring optimal fermentation progress and product quality [18].
Table 2: CRISPR Biosensor Performance in Dairy Applications
| Target | CRISPR System | Amplification | Readout | Limit of Detection (LOD) | Reference |
|---|---|---|---|---|---|
| Listeria spp. | Cas12a | RPA | Fluorescence | aM (attomolar) range | [18] |
| E. coli O157:H7 | n.s. | n.s. | Electrochemical (Microelectrode) | 20 minutes (total assay time) | [18] |
| Lactobacillus plantarum | n.s. | n.s. | Calorimetric | 4.7 - 18.6 hours | [18] |
Meat Fermentation and Safety
The meat production chain, from farming to packaging, benefits from CRISPR biosensors in monitoring spoilage and pathogenic bacteria.
- Spoilage Monitoring: Biosensors can detect spoilage organisms like Pseudomonas by targeting species-specific genes or associated volatile amines, providing an early warning of quality degradation [18] [96].
- Pathogen Detection: CRISPR platforms have been successfully configured to detect major meat-borne pathogens such as Salmonella and Campylobacter. For instance, an electrochemical CRISPR/Cas12a biosensor integrated with saltatory rolling circle amplification (SRCA) achieved ultrasensitive detection of Salmonella Typhimurium [50] [96].
- Starter Culture Synergy: In fermented meat products like sausages, biosensors can monitor the synergistic interaction between starter cultures like Lactobacillus spp. and Staphylococcus spp. by detecting volatile compounds indicative of successful fermentation [18].
Plant Fermentation and Precision Ingredient Production
Precision fermentation uses engineered microbes to produce specific food ingredients, and CRISPR biosensors are key tools for process optimization and quality control.
- Strain Engineering Verification: CRISPR-Cas9 is used to engineer microbial strains (e.g., Komagataella phaffii, Bacillus subtilis) for synthesizing proteins like casein and whey. Biosensors can confirm successful gene edits and monitor the expression of these target products during fermentation [103] [104].
- Pathogen Screening in Cultivated Meat: In the production of cell-based meats, biosensors can screen for potential microbial contaminants, such as Mycoplasma, during the cell culture process, ensuring the safety of the final product [96].
- Metabolite Monitoring: AI-driven models are being developed to predict the output of key compounds. For example, generative adversarial networks (GANs) have predicted ester-synthesis pathways in yeast, and biosensors could be deployed to monitor these flavor-active metabolites in real-time [103].
Experimental Protocols
Protocol: DetectingSalmonella spp.in Meat Using Cas12a-Fluorescence
This protocol outlines the steps for detecting Salmonella genomic DNA using a Cas12a-based biosensor with fluorescent readout, adapted from published platforms like DETECTR [79] [102] [50].
1. Sample Preparation and Nucleic Acid Extraction
- Sample: Homogenize 25 g of meat sample in 225 mL of enrichment broth.
- Enrichment: Incubate at 37°C for 16-18 hours to increase bacterial load.
- Extraction: Extract genomic DNA from 1 mL of enriched culture using a commercial DNA extraction kit. Elute DNA in 50-100 µL of nuclease-free water. Measure DNA concentration and purity spectrophotometrically.
2. Recombinase Polymerase Amplification (RPA)
- Purpose: To amplify a Salmonella-specific gene sequence (e.g., the invA gene) isothermally for high sensitivity.
- Reaction Setup (50 µL):
- 2 µL template DNA
- 29.5 µL rehydration buffer (from kit)
- 10 µL of each forward and reverse primer (10 µM)
- 1 µL magnesium acetate (from kit)
- Add nuclease-free water to 49.5 µL
- Add one RPA pellet and dissolve thoroughly
- Incubation: 39°C for 15-20 minutes.
- Post-amplification: Use 2 µL of the RPA product directly in the CRISPR detection step.
3. CRISPR-Cas12a Detection
- Reaction Mix (20 µL):
- 2 µL of RPA product
- 100 nM LbCas12a enzyme
- 120 nM of Salmonella-specific crRNA
- 1X NEBuffer 2.1
- 500 nM of ssDNA Fluorescent Reporter (e.g., 5'-6-FAM-TTATT-BHQ1-3')
- Add nuclease-free water to 20 µL
- Incubation: 37°C for 10-30 minutes. Protect from light.
4. Signal Readout
- Fluorometer: Transfer the reaction mix to a cuvette and measure fluorescence (Excitation: 485 nm, Emission: 528 nm).
- Real-time PCR Machine: Monitor fluorescence in real-time for kinetic analysis.
- Lateral Flow Dipstick: As an alternative, use an ssDNA reporter labeled with FAM and biotin. After CRISPR reaction, apply the mix to a lateral flow strip. A positive test shows two lines (control and test).
Protocol: MonitoringLactobacillusStarter Cultures via dCas9-Based Electrochemical Sensing
This protocol utilizes catalytically inactive Cas9 (dCas9) for the specific capture and label-free detection of LAB DNA [102] [50].
1. Electrode Functionalization
- Electrode: Clean a gold screen-printed electrode (SPE) electrochemically.
- Probe Immobilization: Incubate the electrode with 1 µM thiol-labeled ssDNA probe complementary to the target Lactobacillus DNA region for 1 hour. The probe forms a self-assembled monolayer on the gold surface.
- Blocking: Rinse the electrode and incubate in 1 mM 6-mercapto-1-hexanol for 30 minutes to block non-specific binding sites.
2. Sample Hybridization and dCas9 Binding
- DNA Denaturation: Heat the extracted DNA sample (from fermentation broth) to 95°C for 5 minutes and immediately cool on ice to obtain single-stranded DNA.
- Hybridization: Incubate 10 µL of denatured DNA with the functionalized electrode at 37°C for 30 minutes. Target DNA will hybridize with the immobilized probe.
- dCas9/sgRNA Binding: Incubate the electrode with a solution containing 200 nM dCas9 protein complexed with a target-specific sgRNA for 20 minutes at room temperature. The dCas9/sgRNA complex will bind to the hybridized dsDNA.
3. Electrochemical Measurement
- Method: Use Electrochemical Impedance Spectroscopy (EIS) or Differential Pulse Voltammetry (DPV).
- Procedure: Measure the electrochemical signal in a solution containing 5 mM [Fe(CN)₆]³⁻/⁴⁻. The binding of the large dCas9/sgRNA complex to the electrode surface increases the electron transfer resistance (Rₑₜ), which is quantitatively measured.
- Quantification: The change in Rₑₜ is proportional to the amount of target DNA captured on the electrode.
The workflow for this electrochemical detection method is illustrated below.
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Reagents for CRISPR-Based Biosensing in Food Fermentation
| Reagent / Material | Function / Role | Example / Note |
|---|---|---|
| Cas Proteins | Core effector enzyme for target recognition and cleavage. | LbCas12a, AapCas12b, LwCas13a; choice depends on target (DNA/RNA) [79] [50]. |
| Synthetic crRNA | Guides the Cas protein to the specific target nucleic acid sequence. | Must be designed for each unique target; critical for specificity [79] [102]. |
| Isothermal Amplification Kits | Pre-amplifies target nucleic acid to enhance detection sensitivity. | Recombinase Polymerase Amplification (RPA) or LAMP kits [79] [50]. |
| Fluorescent Reporter Probes | ssDNA/RNA reporters that generate signal upon trans-cleavage. | FAM-TTATT-BHQ1 (for Cas12); FAM-UUUU-BHQ1 (for Cas13) [102]. |
| Electrochemical Reporters | Redox-active molecules for signal transduction on electrodes. | Methylene Blue-labeled ssDNA; Ferricyanide redox couple [50]. |
| Screen-Printed Electrodes (SPEs) | Disposable, miniaturized platforms for electrochemical detection. | Gold, carbon, or graphene working electrodes [50]. |
| Nucleic Acid Extraction Kits | Isolates high-quality DNA/RNA from complex food matrices. | Must efficiently remove PCR inhibitors from food samples. |
| Lateral Flow Dipsticks | Provides a simple, equipment-free visual readout. | Used with FAM/biotin-labeled reporters for yes/no results [102]. |
The deployment of CRISPR biosensors in dairy, meat, and plant fermentation marks a significant leap forward for food safety and process control. These tools provide researchers and industry professionals with unprecedented capabilities for rapid, specific, and sensitive detection of microbial targets, moving diagnostics from the central lab to the production line. As these technologies continue to mature—through integration with nanotechnology, artificial intelligence, and improved electronics—their role in building a more transparent, efficient, and safe food supply chain is poised to expand dramatically [18] [50] [103].
The Road to Standardization and Regulatory Approval for Widespread Industry Adoption
The integration of CRISPR-based biosensors into food fermentation research represents a paradigm shift in microbial detection and process control. These systems merge the exceptional programmability and specificity of CRISPR-Cas systems with the sensitivity and portability of biosensors, enabling real-time monitoring of microbial populations in complex fermentation matrices [63] [5]. For researchers and drug development professionals, this technology offers unprecedented capabilities for tracking starter cultures, detecting contaminating pathogens, and characterizing microbial interactions that define product quality and safety [18].
Despite their transformative potential, the path from laboratory validation to widespread industry adoption is contingent upon establishing robust standardization frameworks and securing regulatory approval. Current research demonstrates that CRISPR-based detection platforms, particularly those utilizing Cas12 and Cas13 effectors, can achieve sensitivity comparable to PCR (with limits of detection as low as 10 CFU/g for pathogens like Listeria in dairy products) while offering significantly faster results and field-deployability [34] [5]. However, transitioning these proof-of-concept successes into approved, standardized tools requires systematic addressing of performance validation, regulatory alignment, and implementation challenges specific to food fermentation environments.
Core Mechanisms of CRISPR-Based Detection
CRISPR-based biosensors function through a two-step mechanism: target recognition and signal transduction. The process begins with the complementary pairing of CRISPR RNA (crRNA) with target nucleic acids (DNA or RNA) from microbial populations in the fermentation sample [34]. Upon successful binding, Cas proteins such as Cas12a (targeting DNA) or Cas13a (targeting RNA) undergo conformational changes that activate their collateral, trans-cleavage activity [34] [5]. This nonspecific nuclease activity cleaves nearby reporter molecules (e.g., fluorescent or electrochemical probes), generating a measurable signal that confirms pathogen presence or microbial identity [63] [34].
The following diagram illustrates this core detection mechanism:
Key Applications in Food Fermentation Research
CRISPR-based biosensors address multiple critical needs in fermentation monitoring and control:
- Starter Culture Tracking: Real-time monitoring of defined starter cultures (e.g., Lactococcus, Lactobacillus, Streptococcus species) to ensure fermentation initiation and progression [18].
- Pathogen and Contaminant Detection: Rapid identification of spoilage microorganisms (e.g., Pseudomonas) and pathogens (e.g., Listeria monocytogenes, Escherichia coli O157:H7) within 20 minutes to a few hours, significantly faster than culture-based methods [18] [5].
- Process Optimization: Dynamic monitoring of microbial succession and population dynamics enables data-driven decisions to optimize fermentation parameters and terminate processes at optimal quality points [26].
Table 1: Performance Metrics of CRISPR-Based Biosensors for Microbial Targets Relevant to Food Fermentation
| Target Microorganism | CRISPR System | Detection Mechanism | Reported Sensitivity | Time to Result |
|---|---|---|---|---|
| Listeria monocytogenes | Cas12a | Electrochemical | 10 CFU/g [105] | <4 hours [63] |
| E. coli O157:H7 | Cas12a | Fluorescence (DETECTR) | 1-10 CFU/g [5] | 20 min [18] |
| Salmonella spp. | Cas13 | Electrochemical | ~50 CFU/mL [63] | ~2 hours [18] |
| Lactic Acid Bacteria | Cas9 | Colorimetric (SHERLOCK) | ~100 copies/μL [34] | ~1 hour [26] |
| Fungal Contaminants | Cas12 | Lateral Flow | 10-100 spores/g [5] | ~30 minutes [5] |
Experimental Protocols for CRISPR-Based Detection in Fermentation Matrices
Protocol 1: Detection of Bacterial Pathogens in Fermented Dairy Products
This protocol details the steps for detecting bacterial pathogens such as Listeria monocytogenes in yogurt, kefir, and cheese using a Cas12a-based electrochemical biosensor, adapted from published methodologies [63] [18].
Sample Preparation:
- Sample Collection: Aseptically collect 25g of fermented dairy product into a sterile stomacher bag.
- Enrichment (if needed): Add 225mL of appropriate enrichment broth (e.g., UVM modified Listeria enrichment broth) and homogenize for 60s. Incubate at 30°C for 4-6 hours to increase target pathogen concentration while suppressing background flora.
- Nucleic Acid Extraction: Extract DNA using a commercial kit or rapid lysis protocol:
- Transfer 1mL of enriched sample to a microcentrifuge tube
- Centrifuge at 12,000 × g for 2 minutes
- Resuspend pellet in 200μL of lysis buffer (20mM Tris-HCl, 2mM EDTA, 1.2% Triton X-100, 20mg/mL lysozyme)
- Incubate at 37°C for 15 minutes, then heat at 95°C for 5 minutes
- Centrifuge at 12,000 × g for 2 minutes, collect supernatant containing DNA
Recombinase Polymerase Amplification (RPA):
- Prepare RPA Master Mix:
- 29.5μL rehydration buffer
- 2.4μL forward primer (10μM)
- 2.4μL reverse primer (10μM)
- 5μL DNA template
- 10.7μL nuclease-free water
- Add Magnesium Acetate:
- Transfer 47.5μL of master mix to RPA tube
- Add 2.5μL magnesium acetate (280mM) to tube lid
- Centrifuge briefly to combine
- Amplify:
- Incubate at 39°C for 15-20 minutes
CRISPR-Cas12a Detection:
- Prepare CRISPR Reaction:
- 5μL NEBuffer 2.1 (or equivalent)
- 2μL crRNA (1μM) targeting Listeria hlyA gene
- 1μL Cas12a enzyme (10μM)
- 1μL ssDNA reporter (5μM) with methylene blue redox tag
- 36μL nuclease-free water
- Combine and Incubate:
- Add 5μL RPA amplicon to CRISPR reaction mix
- Incubate at 37°C for 10 minutes to allow trans-cleavage activity
Electrochemical Measurement:
- Transfer 50μL of reaction mixture to electrochemical cell
- Apply square wave voltammetry from -0.5V to 0.2V
- Measure current decrease relative to negative control
- Interpret signal reduction >30% as positive detection
Protocol 2: Monitoring Starter Culture Viability in Meat Fermentation
This protocol enables real-time tracking of starter culture viability during sausage and other meat fermentations using a Cas13a-based RNA detection system [18] [26].
Sample Processing:
- Sample Collection: Aseptically collect 10g of fermenting meat product into 90mL peptone water
- Homogenize using a stomacher or vortex mixer for 2 minutes
- Centrifuge at 500 × g for 2 minutes to remove particulate matter
- Collect supernatant and centrifuge at 12,000 × g for 5 minutes to pellet microbial cells
- RNA Extraction:
- Resuspend pellet in 200μL TRIzol reagent
- Incubate 5 minutes at room temperature
- Add 40μL chloroform, shake vigorously for 15 seconds
- Centrifuge at 12,000 × g for 15 minutes at 4°C
- Transfer aqueous phase to new tube
- Precipitate RNA with 100μL isopropanol
- Wash pellet with 75% ethanol, air dry, and resuspend in 20μL nuclease-free water
Reverse Transcription RPA (RT-RPA):
- Prepare RT-RPA Mix:
- 29.5μL rehydration buffer
- 2.4μL forward primer (10μM)
- 2.4μL reverse primer (10μM)
- 2μL RNA template
- 0.5μL reverse transcriptase (100U/μL)
- 7.7μL nuclease-free water
- Add Magnesium Acetate and amplify as in Protocol 1
CRISPR-Cas13a Detection:
- Prepare CRISPR Reaction:
- 5μNEBuffer r3.1 (or equivalent)
- 2μL crRNA (1μM) targeting species-specific 16S rRNA
- 1μL Cas13a enzyme (10μM)
- 1μL RNA reporter (5μM) with FAM-BHQ quenched fluorophore
- 36μL nuclease-free water
- Combine and Incubate:
- Add 5μL RT-RPA amplicon to CRISPR reaction mix
- Incubate at 37°C for 15 minutes
- Measure Fluorescence:
- Transfer to quartz cuvette or microplate reader
- Excite at 495nm, measure emission at 520nm
- Calculate signal-to-noise ratio (positive > 3:1)
The complete workflow for microbial detection in fermentation samples is illustrated below:
Standardization Framework and Validation Requirements
Analytical Performance Standards
For CRISPR-based biosensors to achieve regulatory approval, they must demonstrate consistent performance against established benchmarks. The following table outlines key validation parameters and target performance criteria:
Table 2: Standardization Requirements for CRISPR-Based Biosensors in Food Fermentation Applications
| Performance Parameter | Acceptance Criteria | Validation Method | Reference Method |
|---|---|---|---|
| Analytical Sensitivity (LoD) | ≤10 CFU/g for pathogens | Probit analysis (n=20 replicates) | Culture plating [105] |
| Analytical Specificity | ≥99.5% inclusivity; ≥99% exclusivity | Testing against 50 target and 30 non-target strains | PCR/sequencing [5] |
| Repeatability (Precision) | CV ≤15% for quantitative assays | n=10 replicates across 3 days | Statistical analysis [63] |
| Reproducibility | CV ≤20% between operators/labs | Interlaboratory study (≥3 labs) | ISO 16140-2 [105] |
| Robustness | Function across pH 5.5-8.5, 15-40°C | Deliberate parameter variations | Internal validation [26] |
| Matrix Effects | LoD ≤10 CFU/g in all relevant matrices | Testing in 5+ fermentation matrices | Comparative recovery [63] |
Reference Materials and Controls
Standardized reference materials are essential for method validation and quality control:
- Positive Control Materials: Quantified genomic DNA or synthetic gene fragments for each target pathogen (e.g., hlyA gene for Listeria, stx genes for E. coli O157:H7) with established copy number concentrations [105].
- Matrix-Matched Controls: Fermentation samples (e.g., yogurt, sausage, kimchi) spiked with known concentrations of target microorganisms, processed alongside test samples.
- Process Controls: Non-target microorganisms to verify assay specificity and monitor for cross-reactivity.
The Scientist's Toolkit: Essential Research Reagent Solutions
Table 3: Key Research Reagent Solutions for CRISPR-Based Detection Development
| Reagent/Category | Specific Examples | Function in Workflow | Technical Considerations |
|---|---|---|---|
| CRISPR Enzymes | Cas12a (Cpf1), Cas13a, Cas14 | Sequence-specific detection with collateral cleavage activity | Cas12a for DNA, Cas13a for RNA targets; optimize concentration to minimize non-specific signal [34] |
| crRNA Design | Target-specific crRNA | Guides Cas protein to complementary nucleic acid sequence | Design to target conserved regions with appropriate PAM/ PFS; avoid secondary structures [5] |
| Isothermal Amplification Kits | RPA (TwistAmp), LAMP (Loopamp) | Amplifies target sequences without thermal cycling | RPA: 37-42°C, 15-20 min; LAMP: 60-65°C, 30-60 min; optimize primer design [5] |
| Reporter Molecules | FAM-ssDNA-BHQ1, Methylene Blue-ssDNA | Signal generation via trans-cleavage | Fluorescent for high sensitivity, electrochemical for portability; optimize concentration [63] |
| Nucleic Acid Extraction | Magnetic bead-based kits, Column-based kits | Isolates DNA/RNA from complex fermentation matrices | Evaluate yield, purity, and inhibitor removal; consider rapid lysis for field deployment [18] |
| Signal Detection Platforms | Portable fluorometers, Electrochemical readers, Lateral flow strips | Converts biochemical signal to readable output | Match to application needs: fluorometers for sensitivity, lateral flow for point-of-use [63] [5] |
Regulatory Approval Pathways and Challenges
Current Regulatory Landscape
The regulatory approval pathway for CRISPR-based biosensors in food safety applications involves multiple agencies and frameworks globally. In the United States, the Food and Drug Administration (FDA) and United States Department of Agriculture (USDA) oversee these technologies, while in the European Union, the European Food Safety Authority (EFSA) provides scientific opinions that inform regulatory decisions [63] [105]. The lack of unified international standards creates significant challenges for manufacturers seeking global market access.
Key regulatory considerations include:
- Performance Demonstration: Applicants must provide extensive data demonstrating analytical sensitivity, specificity, robustness, and reliability across intended matrices and conditions [105].
- Standard Reference Method Comparison: New CRISPR-based methods must show equivalent or superior performance compared to established reference methods (e.g., ISO standards, culture-based methods) through independent validation studies [105].
- Quality Management Systems: Manufacturing under ISO 13485 quality systems strengthens regulatory submissions and facilitates conformity assessment.
Addressing Technical and Implementation Barriers
Several technical challenges must be addressed to streamline regulatory approval:
- Matrix Interference: Complex fermentation matrices (e.g., dairy, meat, plant-based) contain fats, proteins, and carbohydrates that can inhibit nucleic acid amplification and CRISPR detection. Sample preparation methods must be optimized and validated for each matrix type [63] [5].
- Multiplexing Capability: Few CRISPR-based biosensors currently support simultaneous detection of multiple targets, limiting efficiency for comprehensive fermentation monitoring. Developing multiplex platforms without cross-reactivity remains a significant technical hurdle [63].
- Quantification Limitations: Most current CRISPR detection systems provide qualitative or semi-quantitative results. Developing truly quantitative approaches is essential for monitoring microbial dynamics during fermentation processes [26].
- Scalability and Automation: Transitioning from laboratory prototypes to scalable, automated systems requires engineering solutions for fluid handling, reaction containment, and signal detection in industrial environments [26].
The following diagram illustrates the key challenges and potential solutions on the path to standardization:
The road to standardization and regulatory approval for CRISPR-based biosensors in food fermentation research, while challenging, is navigable through systematic validation, strategic reagent development, and collaborative engagement with regulatory bodies. The exceptional sensitivity and specificity of these systems, combined with their potential for rapid, on-site deployment, position them as transformative tools for advancing microbial detection and process control in fermentation applications.
Future development should prioritize creating standardized reference materials, establishing performance criteria specific to fermentation matrices, and generating robust validation data across diverse laboratory environments. Additionally, the integration of CRISPR-biosensors with emerging technologies such as artificial intelligence for data interpretation, Internet of Things (IoT) platforms for real-time monitoring, and microfluidics for automated sample processing will further enhance their utility and adoption in industrial fermentation settings [26].
As standardization frameworks mature and regulatory pathways become more defined, CRISPR-based biosensors are poised to transition from research tools to indispensable components of the food fermentation quality control arsenal, ultimately enhancing product safety, quality, and consistency across the industry.
Fermented foods represent a significant sector of the global food industry, relying on complex microbial ecosystems to drive biochemical conversions that determine product yield, quality, and safety. Traditional fermentation monitoring has predominantly depended on physical and chemical parameters (pH, temperature, moisture) that indirectly reflect microbial activity [106]. However, these methods possess an inherent lag time and fail to provide real-time, quantitative data on the specific functional microorganisms governing fermentation processes [107] [106]. This gap becomes particularly critical in spontaneous fermentations, where microbial variability directly translates to inconsistent product quality and economic losses [107] [26].
The emergence of CRISPR-based biosensing technologies offers a revolutionary approach to address these limitations. This gap analysis examines the current state of fermentation monitoring, identifies persistent challenges, and evaluates how CRISPR-Cas systems can bridge these divides through their exceptional specificity, sensitivity, and programmability for detecting microbial biomarkers in complex fermentation matrices.
Current Limitations in Fermentation Monitoring
Technological Gaps in Conventional Methods
Table 1: Limitations of Conventional Microbial Monitoring Techniques in Fermentation
| Method Category | Specific Examples | Key Limitations | Impact on Fermentation Monitoring |
|---|---|---|---|
| Physicochemical Parameters | pH, temperature, starch content [106] | Indirect proxies; lagging indicators; do not quantify microbial drivers [106] | Inability for proactive control; limited predictive value for final product quality |
| Culture-Based Methods | Selective plating, colony counting | Time-consuming (days); misses viable but non-culturable (VBNC) cells [18] | Delayed feedback; incomplete assessment of microbial community |
| Molecular Biology Techniques | Quantitative PCR (qPCR) [106] | Requires expensive instruments; limited detection range; complex sample prep [106] | Not suitable for rapid, on-site monitoring; cost-prohibitive for routine use |
| Omics Technologies | Metagenomics, Metatranscriptomics [107] | Generate population-averaged data; mask functional heterogeneity; complex data analysis [107] | Fails to identify key low-abundance functional contributors; not real-time |
Functional and Operational Challenges
Beyond technical limitations, several functional challenges impede effective fermentation control:
- Inability to Track Functional Heterogeneity: Omics techniques generate population-averaged data, masking the metabolic activity of rare taxa and key functional contributors within low-abundance populations that may critically influence fermentation outcomes [107].
- Lack of Real-Time, On-Site Capability: Most advanced molecular methods require centralized laboratories, sophisticated equipment, and specialized personnel, creating significant delays between sampling and results [18] [26]. This prevents real-time process adjustments during critical fermentation windows.
- Scalability and Standardization Deficits: Traditional fermentation monitoring often relies on sensory evaluation and the experience of master fermenters, methods that are difficult to standardize and scale for industrial production [26].
CRISPR-Based Biosensors: Bridging the Technological Gap
Fundamental Principles and Advantages
CRISPR-Cas systems offer a paradigm shift in fermentation monitoring by providing specific, sensitive, and rapid detection of nucleic acid biomarkers. These systems originate from bacterial adaptive immunity and can be programmed to recognize virtually any DNA or RNA sequence with high specificity [108] [32]. For fermentation monitoring, this translates to several key advantages:
- High Specificity: CRISPR-Cas systems can distinguish between closely related microbial strains, enabling precise tracking of starter cultures or spoilage organisms without cross-reactivity [73].
- Single-Molecule Sensitivity: When combined with pre-amplification techniques, CRISPR-based detection can achieve attomolar (10⁻¹⁸ M) sensitivity, capable of identifying low-abundance functional microorganisms that drive fermentation outcomes [108] [56].
- Rapid Results: The collateral cleavage activity of Cas12 and Cas13 enzymes generates detectable signals within minutes after target recognition, significantly reducing analysis time compared to traditional methods [109] [106].
- Point-of-Need Compatibility: CRISPR biosensors can be integrated with portable detection platforms, including lateral flow assays, microfluidic chips, and smartphone-based readers, enabling real-time monitoring at fermentation facilities [18] [110].
CRISPR Systems for Microbial Detection
Table 2: CRISPR-Cas Systems with Applications in Biosensing
| CRISPR System | Target Type | Collateral Activity | Key Features | Potential Fermentation Applications |
|---|---|---|---|---|
| Cas9 | dsDNA [56] | None [32] | Requires PAM sequence (5'-NGG); precise cis-cleavage [56] | Gene editing of starter cultures; less suitable for detection |
| Cas12a (Cpf1) | dsDNA, ssDNA [56] | ssDNA trans-cleavage [56] [32] | Recognizes T-rich PAM; single RuvC domain [56] | Detection of bacterial species; microbial contamination |
| Cas13a (C2c2) | ssRNA [56] | ssRNA trans-cleavage [56] [32] | No PAM requirement; HEPN domain [56] | Monitoring metabolic activity via gene expression |
| Cas14 (Cas12f) | ssDNA [32] | ssDNA trans-cleavage [32] | Very small size (~400-700 aa); no PAM requirement [32] | Potential for compact biosensing devices |
Case Study: Quantitative Monitoring of Baijiu Fermentation
Experimental Protocol: CRISPR-Cas12a Detection ofAcetilactobacillus jinshanensis
A recent groundbreaking study demonstrated the application of a CRISPR-Cas12a-based visual biosensor for monitoring Baijiu fermentation quality by quantifying Acetilactobacillus jinshanensis, a dominant functional microorganism [106].
Protocol: CRISPR-Cas12a-coupled Gold Nanoparticle Visual Biosensor
Sample Preparation:
- Collect Jiupei (fermentation grains) samples from different stages of Jiang-flavour Baijiu fermentation.
- Extract genomic DNA using standard commercial kits.
- Amplify target 16S rDNA sequence of A. jinshanensis (230 bp fragment) using isothermal amplification (recommended: LAMP or RPA).
CRISPR-Cas12a Reaction:
- Prepare reaction mixture containing:
- 5 μL of NEBuffer r2.1
- 2 μL of LbCas12a enzyme (100 nM final concentration)
- 2 μL of crRNA (custom-designed for A. jinshanensis 16S rDNA, 100 nM)
- 2 μL of ssDNA-FQ reporter (500 nM)
- 4 μL of nuclease-free water
- Add 5 μL of amplified DNA sample to the reaction mixture.
- Incubate at 37°C for 30 minutes to allow Cas12a activation and collateral cleavage.
Signal Detection (Dual-Mode): Fluorescence Measurement:
- Measure fluorescence intensity using a plate reader (excitation/emission: 485/535 nm).
- Quantify target concentration based on fluorescence intensity relative to standards.
Gold Nanoparticle Visual Detection:
- Functionalize AuNPs with thiolated DNA probes complementary to uncleaved reporter.
- Add CRISPR reaction product to AuNP solution.
- Observe color change: red (negative) to blue (positive) due to Cas12a-mediated prevention of AuNP aggregation.
- Quantitative analysis via UV-Vis spectrophotometry measuring absorbance ratio (A520/A620).
Data Analysis:
- Construct standard curve using plasmid DNA with target sequence.
- Calculate A. jinshanensis concentration in samples from fluorescence values or absorbance ratios.
- Correlate microbial concentration with fermentation quality parameters.
Performance Metrics and Validation
The CRISPR-Cas12a biosensor demonstrated exceptional performance in fermentation monitoring:
- Sensitivity: Achieved a detection limit of 1 copy/μL for A. jinshanensis, surpassing the sensitivity of conventional qPCR methods [106].
- Quantitative Correlation: Established a linear relationship (R² = 0.845) between A. jinshanensis concentration and base liquor quality, enabling predictive quality assessment [106].
- Discriminatory Power: Significant differentiation between excellent-grade (average fluorescence: 4838) and ordinary-grade workshops (average fluorescence: 2145) [106].
- Specificity: Successfully distinguished A. jinshanensis from other lactic acid bacteria and common fermentation microorganisms without cross-reactivity [106].
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Reagents for CRISPR-Based Fermentation Monitoring
| Reagent/Category | Specific Examples | Function in Experiment | Implementation Considerations |
|---|---|---|---|
| CRISPR Enzymes | LbCas12a (from Lachnospiraceae bacterium), AacCas12b, LwaCas13a [108] [56] | Sequence-specific nucleic acid recognition and collateral cleavage | Select based on target (DNA/RNA); consider PAM requirements; optimize concentration |
| Guide RNAs | crRNA for Cas12 systems, sgRNA for Cas9 [56] [32] | Programmable recognition element that directs Cas enzyme to target sequence | Design to avoid off-target effects; optimize length (typically 20-25 nt); chemical modifications enhance stability |
| Signal Reporters | ssDNA-FQ probes (for Cas12), ssRNA-FQ probes (for Cas13), functionalized AuNPs [106] [32] | Transduce collateral cleavage into detectable signal (fluorescence, colorimetric) | Fluorophore-quencher pair selection critical for signal-to-noise; AuNP size affects color change |
| Amplification Reagents | LAMP, RPA kits [108] [56] | Pre-amplify target sequences to enhance detection sensitivity | Isothermal methods preferred for point-of-use; optimize to minimize non-specific amplification |
| Nanomaterials | Gold nanoparticles (AuNPs), magnetic beads [108] [106] | Signal enhancement (plasmonic effects), sample separation/concentration | AuNP size (10-50 nm) affects colorimetric response; surface functionalization crucial |
| Portable Detectors | Portable fluorimeters, lateral flow strips, personal glucose meters [18] [110] | Enable field-deployable quantitative readouts | Integration with microfluidics enables automation; smartphone cameras as detectors |
Unmet Needs and Future Perspectives
Despite promising advances, several challenges remain in fully realizing the potential of CRISPR-based biosensors for fermentation monitoring:
- Matrix Complexity: Fermentation samples often contain inhibitors that can interfere with CRISPR reactions, necessitating robust sample preparation methods [18].
- Quantification Standardization: Developing universal standards and calibrators for absolute quantification across different fermentation systems and microbial targets [106].
- Multiplexing Capability: Current systems primarily detect single targets; future platforms must evolve to simultaneously monitor multiple microbial targets to capture community dynamics [107].
- Regulatory and Validation Frameworks: Establishing standardized protocols and regulatory pathways for industry adoption, particularly for small-scale traditional producers [26].
- Integration with Smart Technologies: Combining CRISPR biosensors with IoT devices, AI-driven analytics, and automated fermentation control systems for closed-loop bioprocessing [26].
The convergence of CRISPR-based diagnostics with other emerging technologies—including microfluidics, nanomaterials, and artificial intelligence—promises to revolutionize fermentation monitoring by providing unprecedented resolution into microbial dynamics, ultimately enabling predictive control of fermentation processes and consistent production of high-quality fermented foods.
Conclusion
CRISPR-based biosensors represent a paradigm shift in microbial detection for food fermentation, offering a powerful combination of high specificity, sensitivity, and potential for decentralized, real-time monitoring. This synthesis of foundational knowledge, methodological applications, optimization strategies, and comparative validation underscores their capacity to outperform traditional methods in speed and portability, aligning with the WHO's ASSURED criteria for ideal point-of-care tools. Future directions that will fully unlock this potential include the deeper integration of AI for predictive assay design, the development of universal 'plug-and-play' platforms, and the creation of robust, standardized protocols for regulatory acceptance. For biomedical and clinical research, the advancements in multiplexing, handling complex samples, and creating stable, field-deployable diagnostics directly translate to improved tools for outbreak response, antimicrobial resistance surveillance, and personalized medicine, thereby solidifying the role of CRISPR diagnostics at the intersection of food safety and global public health.
References
- 1. CRISPR-Cas systems: prokaryotes upgrade to adaptive ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4025954/]
- 2. CRISPR-Cas immunity in prokaryotes [https://www.nature.com/articles/nature15386]
- 3. CRISPR-Cas Systems: Prokaryotes Upgrade to Adaptive ... [https://www.sciencedirect.com/science/article/pii/S1097276514002160]
- 4. CRISPR-Cas Systems: A Functional Perspective and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12026748/]
- 5. CRISPR revolution: Unleashing precision pathogen ... [https://www.sciencedirect.com/science/article/pii/S104620232500115X]
- 6. Recent Advances in CRISPR/Cas System-Based ... [https://www.mdpi.com/2072-666X/15/11/1329]
- 7. Recent Advances in CRISPR/Cas System-Based ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11596558/]
- 8. A CRISPR/Cas12a biosensor for portable and accessible ... [https://www.sciencedirect.com/science/article/abs/pii/S0956566325004853]
- 9. CRISPR Guide [https://www.addgene.org/guides/crispr/]
- 10. how conformational regulation of CRISPR-Cas effectors ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12599258/]
- 11. Insights into the Mechanism of CRISPR/Cas9-Based Genome ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9850488/]
- 12. An updated evolutionary classification of CRISPR–Cas ... [https://www.nature.com/articles/s41564-025-02180-8]
- 13. Structures, mechanisms and applications of RNA-centric ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11375968/]
- 14. Components of CRISPR/Cas9 [https://blog.addgene.org/components-of-crispr/cas9-our-new-crispr-101-ebook]
- 15. Structure of the type V-C CRISPR-Cas effector enzyme - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC9522604/]
- 16. Mechanisms and engineering of a miniature type V-N ... [https://www.nature.com/articles/s41467-025-61290-3]
- 17. Structural basis for the activation of a compact CRISPR- ... [https://www.nature.com/articles/s41467-023-41501-5]
- 18. Biosensors in Microbial Ecology: Revolutionizing Food ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12300841/]
- 19. CRISPR/Cas12a-Based Biosensing - PubMed Central - NIH [https://pmc.ncbi.nlm.nih.gov/articles/PMC12190445/]
- 20. Signal Amplification by the trans-Cleavage Activity of CRISPR ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9835055/]
- 21. Enhancement of trans-cleavage activity of Cas12a with ... [https://www.nature.com/articles/s41467-020-18615-1]
- 22. CRISPR-based diagnostics | Nature Biomedical Engineering [https://www.nature.com/articles/s41551-021-00760-7]
- 23. Amplification-free CRISPR/Cas based dual-enzymatic ... [https://pubs.rsc.org/en/content/articlehtml/2025/lc/d4lc01039f]
- 24. A Label-Free CRISPR/Cas12a-G4 Biosensor Integrated ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12025128/]
- 25. Innovative Biosensor Uses CRISPR-Cas12a and PER to ... [https://crisprmedicinenews.com/news/innovative-biosensor-uses-crispr-cas12a-and-per-to-quantify-pathogenic-e-coli/]
- 26. Microbial Process Control in Traditional Fermented Foods [https://www.mdpi.com/2311-5637/11/6/323]
- 27. Mechanism and Applications of CRISPR/Cas-9-Mediated ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8388126/]
- 28. The Complete Guide to Understanding CRISPR sgRNA [https://www.synthego.com/guide/how-to-use-crispr/sgrna/]
- 29. CRISPR/Cas fluorescent probes revolutionizing ... [https://www.thno.org/v16p1877.htm]
- 30. Microbial Interactions in Food Fermentation [https://www.mdpi.com/2304-8158/14/14/2515]
- 31. CRISPR-Based Diagnostics: Challenges and Potential ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9872163/]
- 32. Research progress of CRISPR-based biosensors and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9524266/]
- 33. Research progress of CRISPR-based biosensors and ... [https://www.frontiersin.org/journals/bioengineering-and-biotechnology/articles/10.3389/fbioe.2022.986233/full]
- 34. CRISPR‐driven diagnostics: Molecular mechanisms, clinical ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12464570/]
- 35. Frontiers | CRISPR Applications in Food Microbiology: Enhancing... [https://www.frontiersin.org/research-topics/72933/crispr-applications-in-food-microbiology-enhancing-safety-and-quality]
- 36. CRISPR-Cas13a as a next-generation tool for rapid and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12150587/]
- 37. Research progress on the application of RPA-CRISPR/ ... [https://www.frontiersin.org/journals/cellular-and-infection-microbiology/articles/10.3389/fcimb.2025.1640938/full]
- 38. CRISPR-Cas13a system: A novel tool for molecular ... [https://www.frontiersin.org/journals/microbiology/articles/10.3389/fmicb.2022.1060947/full]
- 39. CRISPR–Cas based platforms for RNA detection [https://pubs.rsc.org/en/content/articlehtml/2025/cc/d5cc03257a?page=search]
- 40. CRISPR-Cas12 Application for the Detection of ... [https://www.mdpi.com/1422-0067/26/17/8732]
- 41. Research progress on the application of RPA-CRISPR ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12343600/]
- 42. A Review of Isothermal Amplification Methods and Food- ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8833899/]
- 43. Comparative Evaluation of PCR-Based, LAMP and RPA ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11172161/]
- 44. PAM-free CRISPR/Cas12a biosensor for PNAs-assisted ... [https://pubmed.ncbi.nlm.nih.gov/40972923/]
- 45. A Real-Time Recombinase Polymerase Amplification ... [https://www.frontiersin.org/journals/microbiology/articles/10.3389/fmicb.2020.586981/full]
- 46. Light-controlled one-pot RPA-CRISPR/Cas12a biosensor ... [https://www.sciencedirect.com/science/article/abs/pii/S0925400525017010]
- 47. A Reverse Transcription-free CRISPR/Cas12a Biosensor ... [https://www.sciencedirect.com/science/article/abs/pii/S0039914025015632]
- 48. Exploring the diagnostic synergy of isothermal amplification ... [https://pubs.rsc.org/en/content/articlehtml/2025/sd/d5sd00080g]
- 49. Flexible Biosensors Based on Colorimetry, Fluorescence ... [https://www.frontiersin.org/journals/bioengineering-and-biotechnology/articles/10.3389/fbioe.2021.753692/full]
- 50. Recent advances in electrochemical-based CRISPR/Cas ... [https://www.sciencedirect.com/science/article/abs/pii/S0963996925015510]
- 51. Electrochemical lateral flow assays: A new frontier for rapid ... [https://www.sciencedirect.com/science/article/pii/S2451910325001097]
- 52. Enhanced detection of Listeria monocytogenes using ... [https://www.sciencedirect.com/science/article/abs/pii/S0003267023011261]
- 53. Rapid Nucleic Acid Detection of Listeria monocytogenes ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10971093/]
- 54. A Portable, Visual, and Dual-Mode Biosensor for... | SpringerLink [https://link.springer.com/protocol/10.1007/978-1-0716-4358-7_16]
- 55. Rapid and Sensitive Detection of Salmonella spp. Using ... [https://www.frontiersin.org/journals/microbiology/articles/10.3389/fmicb.2021.732426/full]
- 56. Recent Advances in the CRISPR/Cas-Based Nucleic Acid ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11507162/]
- 57. /Cas- CRISPR colorimetric based : a promising tool for the... biosensors [https://pubs.rsc.org/en/content/articlelanding/2024/ay/d4ay00578c]
- 58. Intelligent Biosensors Promise Smarter Solutions in Food ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10815208/]
- 59. CRISPR–Cas systems as emerging tools for precision ... [https://www.sciencedirect.com/science/article/abs/pii/S0963996925021416]
- 60. Recent Advances in the CRISPR/Cas-Based Nucleic Acid ... [https://www.mdpi.com/2304-8158/13/20/3222]
- 61. Developing CRISPR-based electrochemical biosensor ... [https://www.sciencedirect.com/science/article/abs/pii/S0889157525001280]
- 62. Interrelations Between Food Form, Texture, and Matrix ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9174310/]
- 63. CRISPR/Cas-Powered Electrochemical Sensors for Food ... [https://www.sciencedirect.com/science/article/abs/pii/S0924224425005321]
- 64. Exploring the Power of the Food Matrix in Health Outcomes [https://dairycouncilofca.org/news-media/exploring-the-power-of-the-food-matrix-in-health-outcomes]
- 65. The effect of fat content in food matrix on the structure, ... [https://www.sciencedirect.com/science/article/abs/pii/S0268005X21008808]
- 66. Magnetic relaxation switching biosensor based on CRISPR ... [https://www.sciencedirect.com/science/article/abs/pii/S030438942502583X]
- 67. Magnetic relaxation switching biosensor based on CRISPR ... [https://pubmed.ncbi.nlm.nih.gov/40865226/]
- 68. Advances in the application of CRISPR technology ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12620484/]
- 69. A Label-Free CRISPR/Cas12a-G4 Biosensor Integrated ... [https://www.mdpi.com/2079-6374/15/4/230]
- 70. Powerful CRISPR-Based Biosensing Techniques and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8905290/]
- 71. Expanding the CRISPR/Cas toolkit: applications in ... [https://www.frontiersin.org/journals/bioengineering-and-biotechnology/articles/10.3389/fbioe.2025.1713700/full]
- 72. CMN Weekly (10 October 2025) [https://crisprmedicinenews.com/news/cmn-weekly-10-october-2025-your-weekly-crispr-medicine-news/]
- 73. Current application and future prospects of CRISPR-Cas in ... [https://www.sciencedirect.com/science/article/abs/pii/S0963996925006520]
- 74. Perspective: Solidifying the impact of cell-free synthetic ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6358126/]
- 75. A Design of Experiments Approach for Enhancing Room ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11939460/]
- 76. Evaluation of a Lyophilized CRISPR-Cas12 Assay for ... [https://pubmed.ncbi.nlm.nih.gov/33807908/]
- 77. Microbial function in food fermentations: Current research ... [https://www.sciencedirect.com/science/article/pii/S266651742500152X]
- 78. Recent advances in the development of food ... [https://www.sciencedirect.com/science/article/abs/pii/S0026265X25035349]
- 79. CRISPR-based Biosensors for Human Health: A Novel ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10571337/]
- 80. CRISPR/Cas12a-based technology: A powerful tool for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8885088/]
- 81. Thermally programmed one-pot CRISPR assay for on-site ... [https://www.nature.com/articles/s41467-025-65193-1]
- 82. Rational Design of CRISPR/Cas12a-RPA Based One-Pot ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9016756/]
- 83. A novel strategy for predicting the quality of a base liquor [https://www.sciencedirect.com/science/article/pii/S2665927125001030]
- 84. High-throughput microfluidic strategy based on RAA ... [https://www.sciencedirect.com/science/article/abs/pii/S0925400522001599]
- 85. Automated CRISPR/Cas9-based genome editing of human ... [https://www.frontiersin.org/journals/bioengineering-and-biotechnology/articles/10.3389/fbioe.2024.1459273/full]
- 86. Fast but accurate: a systematic review and meta-analysis ... [https://www.frontiersin.org/journals/microbiology/articles/10.3389/fmicb.2025.1703247/full]
- 87. Exploring a CRISPR/Cas12a-powered impedimetric ... [https://www.sciencedirect.com/science/article/abs/pii/S0956566325004816]
- 88. A combination of recombinase polymerase amplification ... [https://www.frontiersin.org/journals/cellular-and-infection-microbiology/articles/10.3389/fcimb.2025.1566603/full]
- 89. Bead-based approaches for increased sensitivity and ... [https://www.nature.com/articles/s41551-025-01498-2]
- 90. A colorimetric, photothermal, and fluorescent triple-mode ... [https://jnanobiotechnology.biomedcentral.com/articles/10.1186/s12951-023-02262-x]
- 91. CRISPR-based one-pot detection: A game-changer in ... [https://www.sciencedirect.com/science/article/abs/pii/S0956566325006621]
- 92. CRISPR-Cas-Based Diagnostics in Biomedicine [https://www.mdpi.com/2079-6374/15/10/660]
- 93. Recent advances in CRISPR-based single-nucleotide ... [https://www.nature.com/articles/s43856-025-00933-4]
- 94. Advancements in CRISPR - Based Biosensing for Next-Gen Point of... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9953454/]
- 95. Advancements in CRISPR-Based Biosensing for Next-Gen ... [https://www.mdpi.com/2079-6374/13/2/202]
- 96. Recent Advances in Biosensor Technologies for Meat ... [https://www.mdpi.com/2304-8158/14/5/744]
- 97. CRISPR/Cas-based detection systems – emerging tools for ... [https://www.degruyterbrill.com/document/doi/10.1515/opag-2025-0458/html?lang=en&srsltid=AfmBOopfHO-haA5EyAR6y-0kEDrW66NmgnrWLBmM-eSKT4hhM9tlrUgC]
- 98. CRISPR/Cas-Based Biosensor As a New Age Detection ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9648122/]
- 99. CRISPR-Cas based virus detection - PubMed Central - NIH [https://pmc.ncbi.nlm.nih.gov/articles/PMC8349459/]
- 100. CRISPR-Cas Systems-Based Bacterial Detection [https://pmc.ncbi.nlm.nih.gov/articles/PMC9221980/]
- 101. CRISPR-Cas Systems-Based Bacterial Detection [https://www.mdpi.com/2075-4418/12/6/1335]
- 102. Fluorescence Signal-Readout of CRISPR/Cas Biosensors for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9599699/]
- 103. Precision to plate: AI-driven innovations in fermentation ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12443733/]
- 104. CRISPR and More Delicious [https://ersgenomics.com/crispr-and-more-delicious/]
- 105. Advancing microbial risk assessment: perspectives from ... [https://www.nature.com/articles/s41538-025-00527-3]
- 106. A novel strategy for predicting the quality of a base liquor: visual... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12152550/]
- 107. Microbial function in food fermentations: Current research ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12590229/]
- 108. /Cas-Based Nanobiosensor Using Plasmonic Nanomaterials to... CRISPR [https://link.springer.com/article/10.1007/s13206-024-00183-x]
- 109. CRISPR-powered quantitative keyword search engine in ... [https://www.nature.com/articles/s41467-024-46767-x]
- 110. CRISPR-powered biosensing platform for quantitative ... [https://www.sciencedirect.com/science/article/abs/pii/S0925400523007098]