Genetically Encoded FRET Biosensors: Design Principles, Advanced Applications, and Optimization Strategies
This article provides a comprehensive overview of the latest advancements in genetically encoded FRET biosensors, essential tools for real-time monitoring of biochemical activities in live cells and organisms.
Genetically Encoded FRET Biosensors: Design Principles, Advanced Applications, and Optimization Strategies
Abstract
This article provides a comprehensive overview of the latest advancements in genetically encoded FRET biosensors, essential tools for real-time monitoring of biochemical activities in live cells and organisms. It covers fundamental design principles, including the critical role of spectral overlap, distance (1-10 nm), and orientation between donor and acceptor fluorophores. The content explores cutting-edge methodological applications from multiplexed imaging of signaling pathways to pathogen detection and in vivo brain imaging. A dedicated section addresses common optimization challenges, such as improving dynamic range and specificity, and discusses robust calibration techniques for reliable, quantitative measurements. Synthesizing recent research up to 2025, this resource is tailored for researchers, scientists, and drug development professionals seeking to implement and advance FRET biosensor technology in biomedical research and clinical applications.
The Essential Blueprint: Understanding FRET Biosensor Core Principles and Components
Förster Resonance Energy Transfer (FRET) is a powerful photophysical process describing the non-radiative transfer of energy from an excited donor chromophore to a suitable acceptor chromophore through long-range dipole-dipole coupling [1] [2]. This mechanism, named after German scientist Theodor Förster who established its quantitative theory in 1948, serves as a "spectroscopic ruler" that enables researchers to measure distances and detect interactions at the molecular scale [3] [1]. The exceptional sensitivity of FRET to nanometer-scale distances—typically in the 1-10 nm range—makes it perfectly suited for studying biological phenomena where molecular proximity defines function, including protein-protein interactions, conformational changes in biomolecules, and the activity of cellular signaling pathways [1] [4].
In the context of genetically encoded biosensors, FRET provides an invaluable tool for monitoring dynamic molecular events in living cells and organisms [3] [4]. These biosensors typically incorporate two fluorescent proteins (FPs)—serving as donor and acceptor—linked by a molecular recognition element that changes conformation in response to a specific biochemical or biophysical signal [4]. This design capitalizes on the extreme distance sensitivity of FRET, where even subtle rearrangements within the biosensor induced by target binding or enzymatic activity can produce measurable changes in energy transfer efficiency [5]. The resulting capacity to visualize spatiotemporal patterns of molecular activities under physiological conditions has revolutionized our ability to decipher complex cellular processes, from mechanotransduction to drug-target interactions [4].
Core Theory and Quantitative Framework
The Distance Dependence of FRET
The foundational principle underlying FRET applications is the inverse sixth-power relationship between transfer efficiency and the distance separating the donor and acceptor fluorophores [2]. This relationship is mathematically described by the equation:
Where:
- E = FRET efficiency (fraction of energy transferred)
- r = actual distance between donor and acceptor
- R₀ = Förster radius (characteristic distance for 50% transfer efficiency)
The dramatic influence of distance on FRET efficiency is visually represented in the following diagram, which illustrates the sharp decline in efficiency as the donor-acceptor separation increases beyond the Förster radius:
This distance dependence enables FRET to probe molecular interactions and conformations that are far below the diffraction limit of conventional light microscopy (~250 nm), providing nanometer-scale spatial resolution critical for biosensor design [1].
The Förster Radius (R₀) and its Determinants
The Förster radius (R₀) represents a characteristic parameter for each donor-acceptor pair, defined as the distance at which the FRET efficiency is 50% [1] [2]. This critical distance typically ranges between 4-8 nm for most commonly used FRET pairs, making it ideally matched to the dimensions of biological macromolecules [1]. The value of R₀ is not fixed but depends on several photophysical properties of the fluorophore pair and their environment, as described by the equation:
R₀⁶ = 8.785 × 10⁻⁵ × (κ² × QD × J) / n⁴ [2]
Where the parameters are defined as follows:
Table: Parameters Determining the Förster Radius
| Parameter | Symbol | Description | Typical Range/Value |
|---|---|---|---|
| Orientation Factor | κ² | Describes relative dipole orientation between donor and acceptor | 0-4 (assumed 2/3 for dynamic random averaging) [1] [2] |
| Donor Quantum Yield | QD | Efficiency of donor fluorescence emission | 0-1 (higher values increase R₀) [2] |
| Spectral Overlap Integral | J | Degree of overlap between donor emission and acceptor absorption | Measured in M⁻¹cm⁻¹nm⁴ (larger overlap increases R₀) [1] [2] |
| Refractive Index | n | Optical property of the medium between fluorophores | ~1.4 for biological systems [2] |
The following diagram illustrates how these critical parameters interrelate to determine the overall FRET efficiency in a biosensing context:
For biosensor engineers, careful selection of FRET pairs with appropriate R₀ values is crucial for maximizing dynamic range and sensitivity to the conformational changes they aim to detect [4].
Experimental Measurement of FRET
Methodologies and Protocols
Multiple experimental approaches have been developed to quantify FRET efficiency in biological systems, each with distinct advantages, limitations, and implementation requirements. The choice of method depends on the specific research question, available instrumentation, and desired quantitative rigor.
Table: Comparison of Major FRET Measurement Techniques
| Method | Principle | Key Measurements | Applications | Advantages/Limitations |
|---|---|---|---|---|
| Sensitized Emission (3-Filter FRET) | Measures increased acceptor emission due to FRET [5] | Donor, acceptor, and FRET channel intensities [5] | Live-cell imaging, dynamic processes [5] | Advantages: Fast, live-cell compatible. Limitations: Requires crosstalk corrections [5] [6] |
| Acceptor Photobleaching | Measures donor recovery after acceptor destruction [7] [2] | Donor intensity before and after bleaching [7] | Fixed cells, validation studies [7] | Advantages: Direct efficiency calculation. Limitations: Destructive, single timepoint [7] |
| Fluorescence Lifetime Imaging (FLIM-FRET) | Measures reduced donor lifetime due to FRET [2] | Donor fluorescence lifetime (τ) [2] | Quantitative cellular imaging [2] | Advantages: Independent of concentration. Limitations: Expensive instrumentation [2] |
| Single-molecule FRET (smFRET) | Measures FRET fluctuations at single molecule level [3] [2] | FRET efficiency distributions over time [2] | Molecular heterogeneity, kinetics [3] | Advantages: Reveals subpopulations. Limitations: Technical complexity [3] |
Quantitative Normalization Methods
For intensity-based FRET measurements (particularly sensitized emission), proper normalization is essential for obtaining quantitative, comparable results. Recent advances have addressed limitations of traditional normalization approaches (e.g., NFRET, FRETN) that fail to account for varying acceptor-to-donor expression ratios commonly encountered in live-cell experiments [5]. The QuanTI-FRET method represents a robust framework that introduces correction factors for differential excitation and detection efficiencies, requiring only a sample of known donor:acceptor stoichiometry for calibration [6]. This approach yields absolute FRET values independent of instrument settings or expression levels, making it particularly valuable for biosensor applications where consistent quantification across experiments is essential [6].
The following workflow diagram outlines the key steps in implementing a robust 3-filter FRET measurement and analysis protocol for biosensor validation:
Research Reagent Solutions for FRET Biosensor Development
The successful implementation of FRET-based biosensors requires carefully selected reagents and materials that optimize the key parameters governing energy transfer efficiency.
Table: Essential Research Reagents for FRET Biosensor Development
| Reagent Category | Specific Examples | Function in FRET Biosensors | Design Considerations |
|---|---|---|---|
| Fluorescent Protein Pairs | CFP-YFP, GFP-RFP, ECFP-YPet [4] | Donor and acceptor chromophores | Spectral overlap, quantum yield, photostability, maturation time [4] |
| Linker Domains | Flexible peptide linkers, rigid α-helical spacers | Control basal distance/orientation between FPs | Length, flexibility, protease sensitivity [4] |
| Molecular Recognition Elements | Phosphorylation substrates, ligand-binding domains, force-sensing modules [4] | Transduce target signal into conformational change | Binding affinity, specificity, allosteric properties [4] |
| Calibration Constructs | Tandem FP fusions, covalently linked standards [6] | Quantify FRET efficiency and validate measurements | Known stoichiometry and distance [6] |
| Expression Systems | Plasmid vectors, viral delivery systems | Biosensor delivery to cellular environments | Expression level, cell type specificity, temporal control [4] |
FRET in Genetically Encoded Biosensor Design
Implementation Strategies and Considerations
The integration of FRET mechanisms into genetically encoded biosensors has created powerful tools for visualizing biochemical activities in living systems. These biosensors typically employ a modular design where a sensing domain is flanked by donor and acceptor fluorescent proteins [4]. Upon detection of the target analyte or force, conformational changes in the sensing domain alter the distance and/or orientation between the FPs, thereby modulating FRET efficiency [4]. This general design principle has been successfully applied to create biosensors for diverse targets including ions (Ca²⁺, H⁺), small molecules, kinase activities, GTPase activation, and mechanical forces [3] [4].
Critical performance parameters for FRET biosensors include:
- Dynamic Range: The ratio between maximum and minimum FRET states [4]
- Affinity/Sensitivity: The analyte concentration producing half-maximal response [4]
- Specificity: Selectivity for target versus interfering substances [4]
- Kinetics: Response time to changes in analyte concentration [4]
Optimizing these parameters requires iterative engineering of all biosensor components, with particular attention to the sensing domain properties and FP pairing to maximize the change in FRET efficiency upon target recognition [4].
Advanced Applications in Mechanobiology and Drug Discovery
Recent advances have demonstrated the particular utility of FRET biosensors in mechanobiology, where they enable visualization of molecular-scale forces within living cells [4]. These mechanosensors typically incorporate a force-sensitive peptide or protein domain that undergoes extension or unfolding under mechanical load, thereby altering the distance between attached FPs and modulating FRET efficiency [4]. Such sensors have revealed how cells sense and respond to mechanical cues from their microenvironment, with implications for understanding diseases including atherosclerosis, fibrosis, and cancer [4].
In drug discovery, FRET biosensors support high-throughput screening approaches by providing direct readouts of compound effects on specific signaling pathways in physiologically relevant cellular contexts [4]. The capacity to monitor kinetic responses rather than single endpoint measurements offers advantages for identifying allosteric modulators and characterizing compound mechanism of action [4]. Recent integration with automated imaging systems and analysis pipelines has further enhanced their utility in pharmaceutical applications [3] [4].
The distance-dependent nature of FRET and the well-defined physical principles governing the Förster radius provide a robust foundation for designing genetically encoded biosensors with exquisite sensitivity to molecular events. The quantitative framework established by Förster theory enables rational design of biosensor prototypes, while advanced normalization methods like QuanTI-FRET facilitate precise measurement of FRET efficiency in living systems [6]. As these methodologies continue to evolve alongside improvements in fluorescent protein technology and computational analysis, FRET-based biosensors will undoubtedly maintain their position as indispensable tools for elucidating complex biological processes and accelerating therapeutic development [3] [4].
Förster Resonance Energy Transfer (FRET)-based biosensors are powerful analytical tools that have revolutionized the study of biological processes within living systems. These genetically encoded biosensors function as molecular-scale rulers, enabling researchers to monitor cellular events with high spatiotemporal resolution. Their architecture is fundamentally based on nonradiative energy transfer from an excited donor fluorophore to a nearby acceptor fluorophore through dipole-dipole interactions [8] [3]. This energy transfer occurs only when the donor and acceptor are within close proximity (typically 1-10 nanometers) and exhibits a strong inverse sixth-power dependence on the distance between them [9] [4]. The core design principle involves coupling molecular sensing capability with a measurable fluorescent readout, creating a powerful platform for investigating diverse cellular phenomena including protein-protein interactions, enzyme activities, ion concentration changes, and metabolic signaling pathways [8] [10] [11].
The significance of FRET biosensors in biomedical research stems from their unique advantages over conventional analytical techniques. They offer dynamic visualization capabilities, high sensitivity, strong anti-interference ability, and a unique amplification effect [11]. Furthermore, as genetically encoded tools, they can be specifically targeted to cellular compartments or organelles, enabling long-term imaging studies in cells, tissues, or whole organisms [12]. This technical guide examines the core architectural components of FRET biosensors, their quantitative foundations, and practical considerations for their implementation in drug discovery and basic research.
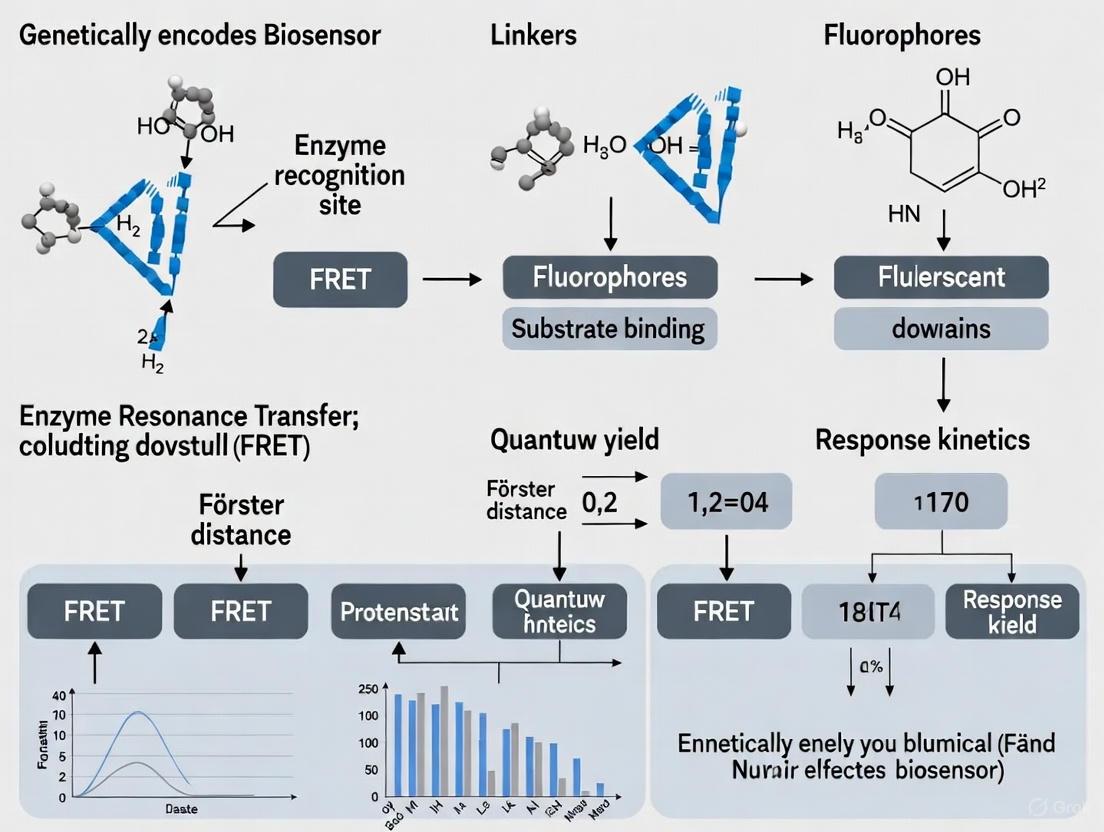
Core Architectural Components
The functional architecture of a genetically encoded FRET biosensor comprises three essential elements: the sensor domain, the ligand domain, and strategically paired fluorescent proteins. The sophisticated integration of these components enables the translation of molecular recognition events into quantifiable fluorescence signals.
Sensor and Ligand Domains
The sensing apparatus of a FRET biosensor consists of complementary protein domains that undergo specific, measurable conformational changes in response to target analytes or environmental conditions. The sensor domain is a biologically derived module that possesses inherent sensitivity to the molecule or condition of interest, such as calcium-binding domains (e.g., calmodulin), phosphorylation sites, or ligand-binding domains [13] [12]. The ligand domain interacts specifically with the sensor domain, and their binding affinity is modulated by the target analyte.
This sensor-ligand interaction directly governs the biosensor's output through several well-established mechanisms:
- Conformational Change: Target binding induces a structural rearrangement in the sensor domain, altering the distance and/or orientation between attached fluorescent proteins (FPs) [13]. This is exemplified by calcium indicators where Ca²⁺ binding to calmodulin (sensor) causes engagement with the M13 peptide (ligand), changing the FP separation [12].
- Cleavage or Separation: Protease activity biosensors incorporate specific cleavage sequences between the FPs. Proteolytic cleavage physically separates the FRET pair, permanently abolishing energy transfer [13].
- Association/Dissociation: Intermolecular FRET biosensors utilize separate sensory and substrate elements fused to different FPs. Ligand-induced association brings the FPs into proximity, enabling FRET [13].
The performance of these sensing systems is characterized by several critical parameters. The dynamic range refers to the total change in FRET ratio between the fully inactive and fully active biosensor states. Gain quantifies the percentage change in FRET ratio following stimulation, while sensitivity defines the analyte concentration required to achieve half-maximal FRET response [4].
Fluorescent Protein Pairs
The selection of appropriate fluorescent protein pairs is crucial for optimizing FRET biosensor performance. Genetically encoded FPs offer the significant advantage of direct genetic fusion to sensor domains, enabling biosensor expression in live cells and organisms without additional labeling steps [11].
The cyan-yellow FP pair, particularly CFP-YFP and their enhanced variants (e.g., ECFP-YPet), represents the most historically common combination in FRET biosensors due to their significant spectral overlap and reliable performance [4] [11]. However, ongoing protein engineering efforts have substantially expanded the palette of available FPs, leading to improved biosensors with different spectral characteristics and enhanced photophysical properties [10] [11].
Table 1: Characteristics of Common Fluorescent Protein Pairs for FRET Biosensors
| Donor | Acceptor | Spectral Overlap | Förster Radius (R₀) | Key Advantages | Common Applications |
|---|---|---|---|---|---|
| CFP | YFP | High | ~4.9-5.2 nm | Well-characterized, reliable | General intracellular signaling [11] |
| mTurquoise2 | cp173Venus | Enhanced | ~5.1 nm | Improved quantum yield, reduced pH sensitivity | High-performance biosensing [10] |
| ECFP | YPet | Very high | ~5.3 nm | High sensitivity for single-cell imaging | Mechanobiological studies [4] |
| GFP-derived mutants | RFP variants | Moderate | Varies | Enables multiplexing with CFP-YFP | Multi-analyte detection [11] |
| BFP | GFP | Limited | Smaller | Minimal spectral cross-talk | Specialized applications [11] |
Beyond traditional CFP-YFP pairs, recent developments have introduced biosensors utilizing red-shifted FPs, which offer advantages including reduced autofluorescence, deeper tissue penetration, and compatibility with optogenetic tools [10] [12]. The ongoing refinement of FP characteristics—including quantum yield, maturation time, photostability, and monomericity—continues to drive improvements in FRET biosensor technology [10] [11].
Quantitative Foundations of FRET
The theoretical framework governing FRET efficiency provides the mathematical foundation for biosensor design and data interpretation. The relationship between intermolecular distance and energy transfer efficiency was first quantified by Theodor Förster, whose equations remain central to modern FRET applications [8] [9] [3].
Fundamental FRET Equations
The efficiency of FRET (E) represents the fraction of excitation energy transferred from donor to acceptor and follows a strong inverse relationship with the sixth power of the distance (r) between fluorophores:
In this equation, R₀ represents the Förster radius—the specific distance at which FRET efficiency is 50%. This parameter is characteristic for each donor-acceptor pair and can be calculated from the photophysical properties of the fluorophores:
R₀ = 0.211 · [κ² · QD · J(λ) · n⁻⁴]¹/⁶ (in Å) [9] [4]
Where the variables and their significance in biosensor design are:
Table 2: Parameters in the Förster Radius Equation
| Parameter | Symbol | Definition | Impact on Biosensor Design |
|---|---|---|---|
| Orientation Factor | κ² | Describes relative dipole alignment | Assumed 2/3 for random rotation; constrained orientation affects E [4] |
| Quantum Yield | QD | Donor emission efficiency | Higher values increase R₀ and potential dynamic range [8] |
| Spectral Overlap Integral | J(λ) | Area under donor emission/acceptor excitation curve | Fundamental determinant of pair compatibility [9] |
| Refractive Index | n | Optical property of medium | Typically ~1.4 for biological systems [8] |
For biosensors containing a single donor and multiple acceptors (n), the FRET efficiency equation modifies to:
E = n · R₀⁶ / (n · R₀⁶ + r⁶) [8] [9]
The spectral overlap integral J(λ) is calculated by integrating the area under the curves where the donor emission spectrum (FD) overlaps with the acceptor absorption spectrum (εA):
J(λ) = ∫ FD(λ) · εA(λ) · λ⁴ dλ [9] [4]
These quantitative relationships enable rational biosensor design by predicting how changes in interfluorophore distance will translate to measurable changes in FRET efficiency. The steep distance dependence (r⁻⁶) makes FRET exquisitely sensitive to molecular-scale distance changes in the 1-10 nm range, perfectly suited for detecting conformational changes in protein-based biosensors [3] [11].
Biosensor Design Workflows and Experimental Protocols
The development and implementation of FRET biosensors follows systematic design principles and validation protocols to ensure reliable performance in biological applications. The process integrates molecular biology, protein engineering, and analytical biochemistry techniques.
Biosensor Construction and Implementation
The initial design phase involves identifying appropriate sensing domains with well-characterized ligand-binding properties and conformational changes. These domains are then fused to selected fluorescent protein pairs using flexible peptide linkers that permit necessary structural rearrangements while minimizing non-specific interactions [13] [4]. Common construction strategies include:
- Single-Chain Intramolecular Sensors: The sensor and ligand domains are connected in a single polypeptide chain flanked by FRET pairs. Ligand binding induces conformational changes that alter FP proximity [13].
- Intermolecular Sensors: Separate sensory and substrate elements are fused to different FPs. Target-induced association brings FPs into proximity for FRET detection [13].
- Cleavage-Based Sensors: Protease substrates are inserted between FRET pairs. Proteolytic cleavage physically separates donor and acceptor, reducing FRET efficiency [13].
Following molecular cloning and expression, biosensor performance is systematically characterized through a multi-stage validation process:
Experimental Protocol: FRET Biosensor Characterization
This protocol outlines the standard procedure for characterizing a newly developed FRET biosensor, from in vitro validation to cellular implementation.
Materials Required:
- Purified biosensor protein or expression plasmid
- Target analyte in purified form
- Appropriate cell line for expression (HEK293, HeLa, or specialized lines)
- Fluorescence spectrometer or confocal microscope with FRET capabilities
- Ligands/inhibitors for specificity testing
Procedure:
In Vitro Spectroscopic Characterization
- Express and purify biosensor protein using standard protein purification methods
- Acquire emission spectra with donor excitation in the presence of varying analyte concentrations
- Calculate FRET ratio (acceptor emission / donor emission) for each condition
- Generate dose-response curve by plotting FRET ratio against analyte concentration
- Determine dynamic range as (Rmax - Rmin)/Rmin × 100%, where Rmax and Rmin are maximum and minimum FRET ratios [4]
Specificity and Selectivity Testing
- Challenge biosensor with structural analogs and related molecules
- Test potential interfering substances present in the biological environment
- Verify minimal response to non-target molecules
Cellular Expression and Localization
- Transfect target cells with biosensor plasmid using appropriate methods (lipofection, electroporation)
- Confirm proper subcellular localization using fluorescence microscopy
- Verify biosensor functionality does not disrupt normal cellular processes
Live-Cell Imaging and Data Acquisition
- Plate cells on appropriate imaging chambers and allow attachment
- Transfer to microscope stage with environmental control (37°C, 5% CO₂)
- Acquire time-lapse images using appropriate filter sets for donor and acceptor channels
- Apply stimulation (chemical, mechanical, or optical) during imaging
- Calculate FRET ratio images and analyze temporal dynamics [4]
Data Analysis and Validation
- Perform background subtraction and correction for spectral bleed-through
- Normalize FRET ratios to baseline values for comparative analysis
- Conduct statistical analysis across multiple cells and experimental replicates
- Validate results using pharmacological inhibitors or genetic manipulations
This comprehensive validation ensures that the biosensor provides accurate, reproducible readouts of biological activity while minimizing artifacts from environmental factors or non-specific interactions.
Advanced Applications in Research and Drug Discovery
FRET biosensors have enabled significant advances across multiple biomedical research domains by providing unprecedented access to molecular-scale events in living systems. Their applications span fundamental biological investigation through drug development pipelines.
Cellular Imaging and Mechanobiology
In cellular imaging, FRET biosensors permit real-time monitoring of intracellular signaling events with high spatiotemporal resolution. Notable applications include:
- Calcium Signaling: The GCaMP series (utilizing cpGFP rather than FRET) and Cameleon FRET biosensors have revolutionized calcium imaging in neuronal systems and other cell types [12].
- Kinase Activity: Src kinase FRET biosensors have revealed mechanosensitive signaling dynamics, showing wave propagation from sites of mechanical stimulation [4].
- Metabolic Monitoring: Biosensors for metabolites like NADP, methionine, and isoleucine enable real-time tracking of metabolic fluxes in living cells [14].
- Mechanotransduction: Genetically encoded tension sensors (GETS) incorporating FRET pairs measure molecular-scale forces across proteins in live cells, revealing how cells sense and respond to mechanical cues [8] [4].
Drug Discovery and High-Throughput Screening
The pharmaceutical industry increasingly incorporates FRET biosensors into drug discovery pipelines due to their sensitivity, specificity, and compatibility with live-cell formats:
- Target Engagement: FRET biosensors can directly report on compound binding to target proteins in physiological environments, providing critical information for lead optimization [10].
- Pathway Modulation: By monitoring downstream signaling events, biosensors reveal functional consequences of drug treatments beyond direct target binding [4].
- High-Content Screening: Multiplexed FRET biosensors enable parallel assessment of multiple pathway activities in single cells, generating rich datasets for compound characterization [10] [4].
- Toxicology Assessment: Biosensors monitoring stress pathways, apoptosis, and mitochondrial function provide early indications of compound toxicity [15].
The implementation of FRET biosensors in microfluidic platforms and paper-based analytical devices further extends their utility for point-of-care diagnostics and therapeutic monitoring [14]. Recent innovations include BRET (bioluminescence resonance energy transfer) variants that eliminate the need for external excitation light, reducing autofluorescence and enabling deeper tissue applications [14].
Research Reagent Solutions
The successful implementation of FRET biosensor technology relies on specialized reagents and tools. The following table outlines essential resources for researchers developing or applying FRET biosensors.
Table 3: Essential Research Reagents for FRET Biosensor Development
| Reagent Category | Specific Examples | Function and Application | Key Characteristics |
|---|---|---|---|
| Fluorescent Protein Pairs | CFP-YFP, mTurquoise2-cp173Venus, ECFP-YPet [10] [4] [11] | FRET signal generation; optimized pairs available from plasmid repositories | Spectral compatibility, brightness, photostability |
| Biosensor Plasmids | GCaMP series (calcium), AMPfret (energy status), LUMABS (antibodies) [14] [12] | Ready-to-use biosensor constructs; available from Addgene and other repositories | Validated performance, modular design |
| Expression Systems | pcDNA, lentiviral, AAV vectors [12] | Biosensor delivery to target cells | Efficient transduction, appropriate tropism |
| Sensing Domains | Calmodulin-M13 (calcium), DHFR (small molecules), LivJ (isoleucine) [3] [14] | Target recognition modules for custom biosensor engineering | Specificity, conformational change upon binding |
| Cell Lines | HEK293, HeLa, primary neuronal cultures | Biosensor expression and validation | Transferability, physiological relevance |
| Imaging Equipment | Confocal microscopes, plate readers with FRET capabilities | Signal detection and quantification | Appropriate filter sets, environmental control |
| Reference Standards | Fluorescent beads, control biosensors [4] | Instrument calibration and experimental controls | Stable fluorescence, well-characterized properties |
The sophisticated architecture of FRET biosensors—integrating carefully selected sensor domains, ligand domains, and fluorescent protein pairs—has established them as indispensable tools for modern biological research and drug development. Their genetically encoded nature enables non-invasive monitoring of molecular events in living systems with exceptional spatiotemporal resolution. The quantitative foundation of FRET provides a robust framework for biosensor design and data interpretation, while ongoing advancements in fluorescent protein technology and sensing strategies continue to expand their applications. As these tools become increasingly sophisticated through integration with nanomaterials, multiplexing approaches, and computational methods, FRET biosensors are poised to deliver even deeper insights into cellular function and accelerate the development of novel therapeutics.
Förster resonance energy transfer (FRET) is a physical phenomenon describing energy transfer between two light-sensitive molecules (chromophores), where an excited donor fluorophore non-radiatively transfers its energy to a nearby acceptor fluorophore through dipole–dipole coupling [2]. The efficiency of this energy transfer is inversely proportional to the sixth power of the distance between donor and acceptor, making FRET extremely sensitive to small changes in distance, typically within the 1-10 nanometer range [8] [2]. This exquisite distance dependence provides the foundation for FRET-based biosensors, which are versatile tools for obtaining insights into various biological processes, including cellular imaging, drug discovery, pathogen detection, and cancer diagnosis [16] [8].
In the context of genetically encoded biosensors, FRET technology provides a powerful tool for visualizing signaling molecules in live cells with high spatiotemporal resolution [17]. The selection of optimal donor-acceptor fluorophore pairs is arguably the most critical aspect of FRET biosensor design, as it directly determines the sensor's brightness, dynamic range, and overall performance [17] [18]. Continued research on biosensor design, donor-acceptor pair optimization, and integration of innovative materials is extending applications of FRET biosensors across health care settings [16] [8].
Fundamental Principles of FRET Pair Selection
Key Photophysical Parameters
The performance of a FRET pair depends on several interlinked photophysical parameters that collectively determine the efficiency of energy transfer. The FRET efficiency (E) is quantitatively described by the equation E = 1/(1 + r⁶/R₀⁶), where r is the distance between donor and acceptor dipoles, and R₀ is the Förster radius - the distance at which FRET efficiency is 50% [17] [2]. The Förster radius itself depends on multiple factors expressed in the equation: R₀⁶ = (9000(ln10)κ²QDJ)/(128π⁵Nₐn⁴), where QD is the quantum yield of the donor, κ² is the orientation factor, J is the spectral overlap integral, n is the refractive index, and Nₐ is Avogadro's number [17] [8] [2].
The spectral overlap integral (J) represents the degree of overlap between the donor emission spectrum and the acceptor absorption spectrum, calculated as J = ∫FD(λ)εA(λ)λ⁴dλ, where FD is the donor emission profile, and εA is the acceptor molar extinction coefficient [8] [2]. This overlap is a fundamental requirement for FRET to occur, with a general guideline that substantial overlap (>30%) is necessary for efficient energy transfer [17]. The relative orientation of the donor and acceptor dipole moments, represented by the orientation factor κ², typically assumes a value of 2/3 corresponding to random orientation, though this assumption requires careful consideration for fluorescent proteins which may not undergo rapid rotational diffusion during their excited state lifetime [17].
Practical Selection Criteria
When selecting FRET pairs for biosensor design, several practical criteria must be considered to ensure optimal performance in biological systems. First, the donor should have a high quantum yield to ensure efficient energy transfer, while the acceptor should possess a high extinction coefficient [19]. Both fluorophores should demonstrate high photostability to withstand excitation light during time-lapse imaging, as differences in photobleaching characteristics can result in false FRET changes [18]. For genetically encoded biosensors using fluorescent proteins, additional considerations include maturation efficiency (the fraction of produced protein that results in a correctly folded protein with a functional chromophore), monomeric behavior to prevent aberrant oligomerization, and minimal sensitivity to environmental changes such as pH or halide concentrations [18].
The effective distance for autofluorescent FP-based FRET pairs is less than 7 nm, resulting in practical maximal FRET efficiencies of 40-55% due to the chromophores being centrally buried in the β-barrel structure with a diameter of about 2.4 nm [17]. The FRET dynamic range, defined as (Emax - Emin)/Emin, where Emin and Emax are the minimum and maximum FRET efficiency of a given biosensor, is essential for detection of cellular events with high sensitivity [17]. Since FRET efficiency and distance are related by a sigmoidal curve with the highest slope at its midpoint, a FRET pair with an R₀ approximating the distance that a given FRET biosensor operates at should be selected to maximize dynamic range [17].
Quantitative Comparison of Common FRET Pairs
The performance of FRET-based biosensors depends significantly on brightness and dynamic range, which are highly dependent on the characteristics of the applied fluorescent proteins [18]. The following tables summarize key parameters for commonly used and recently developed FRET pairs, providing a reference for selecting optimal pairs for biosensor development.
Table 1: Photophysical Properties of Commonly Used FRET Pairs
| FRET Pair | Donor Emission Peak (nm) | Acceptor Absorption Peak (nm) | Förster Radius (R₀ in nm) | FRET Efficiency | Key Applications |
|---|---|---|---|---|---|
| CFP-YFP [17] [20] | 475 [20] | 514 [20] | ~4.9-5.2 [17] | 0.4-0.6 [20] | Kinase activity, calcium imaging [17] |
| GFP-RFP [20] | 509 [20] | 584 [20] | - | 0.3-0.5 [20] | Protein-protein interactions [17] |
| Alexa Fluor 488-Alexa Fluor 594 [20] | 519 [20] | 590 [20] | - | 0.5-0.7 [20] | Immunoassays, fixed cell imaging [21] |
| mTurquoise2-mNeonGreen [18] | 474 [18] | 506 [18] | 5.6 [18] | High [18] | Live-cell imaging, GPCR signaling [18] |
| mTurquoise2-mCherry [18] | 474 [18] | 587 [18] | 4.9 [18] | Moderate [18] | Red-shifted sensors, multiplexing [18] |
Table 2: Performance Characteristics of Selected Fluorescent Protein FRET Pairs
| FRET Pair | Brightness | Photostability | Maturation Efficiency | Monomeric Tendency | pH Sensitivity |
|---|---|---|---|---|---|
| CFP-YFP [17] [18] | Moderate [18] | Low (YFP) [18] | Moderate [18] | High (monomeric variants) [18] | Sensitive (YFP) [18] |
| mTurquoise2-mNeonGreen [18] | High [18] | Good [18] | High [18] | High [18] | Low [18] |
| mTurquoise2-sYFP2 [18] | High [18] | Moderate [18] | High [18] | High [18] | Moderate [18] |
| mTurquoise2-mCherry [18] | Moderate [18] | Good [18] | Moderate [18] | High [18] | Low [18] |
| mTurquoise2-mScarlet-I [18] | Moderate-High [18] | Good [18] | Good [18] | High [18] | Low [18] |
Recent research has systematically evaluated acceptor fluorescent proteins paired with the optimized cyan fluorescent protein mTurquoise2, which has a high quantum yield of 93%, monomeric behavior, and good photostability [18]. The results demonstrate that mNeonGreen is the most efficient acceptor for mTurquoise2 with a Förster radius of 5.6 nm and better photostability than SYFP2 [18]. Among orange and red fluorescent proteins, mCherry and mScarlet-I are the best performing acceptors, with mScarlet-I exhibiting relatively high quantum yield that provides sensitized emission surpassing mCherry in a FRET pair [18].
Experimental Protocols for FRET Pair Validation
Measuring FRET Efficiency in Live Cells
Several methodologies exist for measuring FRET efficiency in biological contexts, each with distinct advantages and limitations. These can be broadly categorized into indirect methods, which involve measurements of FRET efficiency at different states, and direct methods, which directly relate changes in fluorescence intensity to FRET changes [17].
Fluorescence Lifetime Imaging FRET (FLIM-FRET) measures the decrease in donor fluorescence lifetime in the presence of acceptor and is considered one of the most robust methods for FRET quantification [17] [2]. The FRET efficiency can be calculated from the equation E = 1 - τD'/τD, where τD' and τD are the donor fluorescence lifetime in the presence and absence of acceptor, respectively [2]. This method is particularly powerful because fluorescence lifetime is independent of fluorophore concentration and excitation light intensity, providing more reliable quantitative measurements [17].
Sensitized Emission FRET (seFRET) involves measuring the increase in acceptor emission intensity when the donor is excited, which occurs due to FRET [17] [2]. This method requires careful correction for spectral bleed-through (direct excitation of acceptor by donor excitation light and emission of donor into acceptor detection channel) using control samples expressing donor-only and acceptor-only constructs [17]. Although more prone to artifacts than FLIM-FRET, seFRET offers high temporal resolution (millisecond scale), enabling tracking of fast molecular events and compatibility with high-throughput screening [17].
Acceptor Photobleaching FRET (apFRET) determines FRET efficiency by measuring the increase in donor fluorescence after photodestruction of the acceptor [2]. The FRET efficiency is calculated as E = 1 - FDA/FD, where FDA is donor fluorescence before acceptor photobleaching and FD is donor fluorescence after photobleaching [2]. While conceptually simple, this method is destructive and not suitable for live-cell kinetic studies, though it can be performed on most standard fluorescence microscopes without specialized equipment [17] [2].
Table 3: Comparison of FRET Measurement Methods
| Method | Suitable in Live Cells | Temporal Resolution | Measures FRET Efficiency | Key Advantages | Key Limitations |
|---|---|---|---|---|---|
| Spectral Imaging FRET (siFRET) [17] | Yes | Second | Yes | Provides full spectral information | Lower temporal resolution |
| Acceptor Photobleaching FRET (apFRET) [17] [2] | No | Not applicable | Yes | Simple implementation, no specialized equipment required | Destructive, not for live cells |
| FLIM-FRET [17] [2] | Yes | Second* | Yes | Insensitive to concentration, robust quantification | Requires specialized equipment |
| Sensitized Emission FRET (seFRET) [17] | Yes | Millisecond | No | High temporal resolution, suitable for high-throughput | Requires careful correction for bleed-through |
| Polarization-resolved FRET (prFRET) [17] | Yes | Millisecond | No | Can detect homo-FRET | Complex data interpretation |
*under single-photon avalanche photodiodes (SPAD)-based FLIM-FRET imaging [17]
Protocol for Validating Protein Interactions Using FRET in Escherichia coli
The following protocol describes a methodology for detecting protein-protein interactions in the cytoplasm and periplasm of Escherichia coli using FRET, which can be adapted for validation of FRET pairs in bacterial systems [22]:
Construct Design: Genetically fuse proteins of interest to selected donor and acceptor fluorescent proteins using standard molecular biology techniques. Ensure the fluorescent proteins are in the same reading frame as the proteins of interest with flexible linkers between domains.
Sample Preparation:
- Transform constructs into appropriate E. coli strains and culture overnight in selective media.
- Dilute cultures and grow to mid-log phase (OD600 ≈ 0.5-0.7).
- Induce expression with appropriate inducers (e.g., IPTG) if using inducible promoters.
- Harvest cells by centrifugation and resuspend in appropriate buffer (e.g., PBS or growth medium) for analysis.
Fluorescence Measurement:
- For fixed-cell measurements: Fix cells with formaldehyde and glutaraldehyde (FAGA fixative), wash, and resuspend in buffer.
- Transfer samples to appropriate measurement containers (quartz cuvettes for spectrofluorometers or glass-bottomed plates for plate readers).
- Measure fluorescence emission spectra with excitation at both donor and acceptor excitation wavelengths.
- Include control samples (donor-only, acceptor-only, and untransfected cells) for background subtraction and spectral unmixing.
Data Analysis and FRET Calculation:
- Subtract background fluorescence from untransfected cells.
- Perform spectral unmixing using reference spectra from donor-only and acceptor-only samples.
- Calculate FRET efficiency from sensitized emission of the acceptor after donor excitation.
- Validate interactions by comparing FRET efficiency in test samples with appropriate negative controls.
This protocol can be adapted for high-throughput screening in 96-well plates and modified for real-time monitoring of interactions in living cells by omitting the fixation step [22].
Advanced Applications and Research Reagent Solutions
Research Reagent Solutions for FRET Experiments
Table 4: Essential Research Reagents for FRET Biosensor Development
| Reagent Category | Specific Examples | Function in FRET Experiments |
|---|---|---|
| Donor FPs [17] [18] | mTurquoise2, CFP, TFP, GFP | Energy transfer initiators; high quantum yield critical |
| Acceptor FPs [17] [18] | mNeonGreen, SYFP2, mCherry, mScarlet-I | Energy transfer receivers; high extinction coefficient critical |
| Molecular Biology Reagents [22] | Restriction enzymes, ligases, polymerases | Construction of FP fusion constructs |
| Cell Culture Materials [22] | Media, antibiotics, inducters (IPTG) | Expression of FP constructs in cells |
| Fixation Reagents [22] | Formaldehyde, glutaraldehyde | Cell fixation for endpoint measurements |
| Measurement Platforms [22] | Fluorometers, plate readers, fluorescence microscopes | Detection and quantification of FRET signals |
Emerging Applications and Future Directions
FRET biosensors continue to find expanding applications across biomedical research. In cellular imaging, they allow real-time monitoring of intracellular events such as protein-protein interactions, enzyme activities, and ion concentration changes [8]. In drug discovery, FRET biosensors enable evaluation of molecular responses to candidate drugs, making them suitable for high-throughput screening platforms [16] [8]. For pathogen detection, FRET biosensors offer rapid, specific, and sensitive identification of infectious agents by targeting pathogen-specific biomolecules [16] [8]. In cancer diagnosis, they contribute to early-stage detection by sensing tumor-specific biomarkers and alterations in cellular signaling pathways [16] [8].
Recent advances include the development of single-molecule FRET (smFRET), which enables the study of individual molecules and provides insights into molecular dynamics and heterogeneity that are obscured in ensemble measurements [8] [2]. smFRET has been applied to study protein folding, molecular interactions, and conformational changes in nucleic acids at the single-molecule level [8] [20]. Integration of FRET with other imaging modalities, such as FRET-fluorescence lifetime imaging (FRET-FLIM) and FRET-super-resolution microscopy, provides complementary information with enhanced spatial resolution [20].
The application of artificial intelligence and Internet of Things technologies to FRET biosensing represents another emerging frontier, enabling automated analysis, remote monitoring, and enhanced data processing capabilities [3]. Continued research on biosensor design, donor-acceptor pair optimization, and integration of innovative materials such as up-converting nanoparticles and conjugated polymers promises to further extend the applications and performance of FRET biosensors across diverse research and clinical settings [8] [3].
The selection of optimal donor-acceptor fluorophore pairs represents a critical consideration in the design of genetically encoded FRET biosensors. Key factors including spectral overlap, quantum yield, extinction coefficient, photostability, and environmental sensitivity must be carefully balanced to achieve biosensors with high dynamic range, brightness, and reliability in biological systems. While traditional pairs like CFP-YFP remain widely used, newly developed pairs such as mTurquoise2-mNeonGreen offer improved photophysical properties and performance. As FRET technology continues to evolve with advancements in measurement methodologies, reagent development, and emerging applications, thoughtful consideration of spectral parameters in fluorophore pair selection will remain fundamental to biosensor optimization and innovation.
FRET Pair Selection Workflow
FRET Fundamental Principle
Förster Resonance Energy Transfer (FRET) biosensors represent a transformative technology in biomedical research, enabling the direct visualization of molecular events within living systems. This whitepaper delineates the fundamental advantages of genetically encoded FRET biosensors over conventional analytical techniques, with particular emphasis on their superior sensitivity, exceptional specificity, and unique capacity for real-time monitoring in live cells. Within the broader context of biosensor design research, we present quantitative performance comparisons, detailed experimental methodologies, and emerging applications that collectively underscore the transformative potential of FRET technology for advancing drug discovery and fundamental mechanistic studies.
Förster Resonance Energy Transfer (FRET) is a distance-dependent, non-radiative energy transfer process between two chromophores—a donor and an acceptor—that occurs when they are in close proximity (typically 1-10 nm) [11] [3]. This photophysical phenomenon provides a powerful mechanism for monitoring molecular interactions, conformational changes, and biochemical activities within the complex milieu of living cells. Genetically encoded FRET biosensors are engineered by fusing fluorescent proteins (FPs) to specific sensing domains that undergo conformational changes in response to target activation [23] [24]. The core architecture typically sandwiches a sensing unit (e.g., a kinase-specific substrate domain and a phospho-amino acid binding domain) between two FPs that constitute a FRET pair. Upon activation, the conformational rearrangement alters the distance and/or orientation between the donor and acceptor FPs, resulting in a measurable change in FRET efficiency [24].
The significance of FRET technology resides in its ability to provide spatiotemporal resolution of biochemical events that is unattainable with conventional endpoint assays. Traditional techniques such as Western blotting, enzyme-linked immunosorbent assays (ELISA), and mass spectrometry require cell lysis, providing only static snapshots of cellular processes and obliterating crucial information about dynamics and heterogeneity [25] [24]. Furthermore, these methods lack the spatial resolution to monitor signaling events within specific subcellular compartments. FRET biosensors overcome these limitations by enabling non-invasive, continuous monitoring of molecular activities directly in living cells and tissues, preserving native physiological context while providing quantitative readouts with high temporal and spatial fidelity [23] [26].
Sensitivity: Detecting Molecular Events at Ultra-Low Concentrations
The sensitivity of FRET biosensors stems from their ability to detect minute changes in distance between molecular components, effectively translating nanometer-scale movements into quantifiable fluorescence signals. This exceptional sensitivity enables researchers to monitor biological interactions and enzymatic activities at concentrations orders of magnitude lower than conventional techniques can reliably detect.
Quantitative Sensitivity Comparisons
Table 1: Sensitivity Comparison Between FRET Biosensors and Conventional Techniques
| Target/Analyte | FRET Biosensor Performance | Conventional Technique | Performance Comparison |
|---|---|---|---|
| BCR-ABL Kinase Activity | Detected imatinib inhibition at 0.1 μM [25] | Western Blotting | Required 0.5-1 μM for detection [25] |
| SARS-CoV-2 Viral RNA | Limit of detection: 10 copies per reaction [3] [27] | Conventional PCR | Similar sensitivity but FRET enables real-time quantification [3] |
| General Biomolecules | Picomolar to nanomolar sensitivity [3] [27] | Colorimetric Assays, ELISA | Typically micromolar sensitivity [3] [27] |
| Protein-Protein Interactions | Effective at nanomolar concentrations [3] | Co-immunoprecipitation | Requires higher protein concentrations [3] |
The underlying mechanism for this enhanced sensitivity lies in the fundamental physics of FRET. Energy transfer efficiency is inversely proportional to the sixth power of the distance between donor and acceptor fluorophores (E ∝ 1/r⁶), rendering the technique exquisitely sensitive to minute distance changes in the 1-10 nm range [11] [28]. This relationship enables FRET biosensors to detect molecular interactions that would be invisible to conventional techniques, which typically rely on bulk measurements with less pronounced dependence on molecular proximity.
Technological Foundations of Enhanced Sensitivity
The exceptional sensitivity of FRET biosensors is further augmented by several design and methodological considerations. Ratiometric measurements, which calculate the ratio of acceptor to donor emission, inherently correct for variations in biosensor concentration, excitation intensity, and photobleaching, thereby reducing noise and enhancing signal-to-noise ratios [23] [29]. Additionally, advanced detection modalities such as Fluorescence Lifetime Imaging Microscopy (FLIM) measure the reduction in donor fluorescence lifetime resulting from FRET, a parameter that is independent of fluorophore concentration and laser power, thus providing superior quantification in complex biological environments [26] [24]. The engineering of optimized FRET pairs with higher quantum yields, better spectral overlap, and improved photostability has progressively enhanced the dynamic range and sensitivity of these biosensors [26] [30]. Furthermore, the implementation of dark acceptors (e.g., ShadowY, ShadowR) in multiplexed FRET systems eliminates spectral cross-talk, thereby improving sensitivity in multi-parameter experiments [30].
Specificity: Precision in Complex Biological Environments
Specificity in FRET biosensors is engineered through multiple interdependent strategies that collectively ensure precise recognition of target analytes amidst the complex background of cellular components. This molecular precision is achieved through sophisticated protein engineering that combines biological recognition elements with optimized fluorophore pairs.
Molecular Mechanisms of Biosensor Specificity
Table 2: Specificity Mechanisms in FRET Biosensor Design
| Specificity Mechanism | Description | Representative Example |
|---|---|---|
| Domain-Based Recognition | Utilizes specific binding domains (e.g., SH2, FHA1, 14-3-3) that recognize phosphorylated motifs [25] [24] | CrkL substrate with SH2 domain for BCR-ABL sensing [25] |
| Conformational Sensing | Leverages natural conformational changes in full-length proteins upon activation [26] [24] | STATeLight biosensors detecting parallel dimerization [26] |
| Intramolecular Binding | Engineered interaction between phosphorylation site and binding domain within same polypeptide [25] [24] | AKAR series for PKA activity monitoring [24] |
| Orthogonal FRET Pairs | Use of spectrally distinct fluorophore pairs to enable multiplexed detection without cross-talk [30] | mNeonGreen/ShadowY and mScarlet-I3/ShadowR pairs [30] |
The specificity of FRET biosensors is fundamentally rooted in their modular design, which incorporates well-characterized biological components with inherent molecular recognition capabilities. For kinase activity sensors, this typically involves a consensus phosphorylation sequence specific to the target kinase, coupled with a phospho-amino acid binding domain (e.g., FHA1, SH2) that selectively engages with the phosphorylated substrate [24]. This dual requirement—recognition by the kinase followed by binding to the recognition domain—creates a two-step verification process that enhances specificity over conventional phosphorylation assays, which may detect off-target phosphorylation events.
Experimental Validation of Specificity
Rigorous validation protocols are essential to establish biosensor specificity. The Pickles biosensor for BCR-ABL activity exemplifies this approach, having undergone comprehensive specificity testing against an array of kinases, in addition to mutational analysis of critical tyrosine and SH2 domains to confirm that FRET changes resulted specifically from the intended intramolecular binding events [25]. Similarly, the recently developed STATeLight biosensors directly monitor STAT activation through conformational rearrangement from antiparallel to parallel dimers, a mechanism that is inherently specific to the activation process and insensitive to potentially confounding signals from inactive phosphorylated monomers or truncated STAT variants [26]. Furthermore, in multiplexed applications, the use of dark acceptors such as ShadowY and ShadowR eliminates spectral bleed-through, thereby preserving specificity when monitoring multiple targets simultaneously [30].
Real-Time Live-Cell Imaging: Monitoring Dynamics in Physiological Contexts
The capacity to monitor biochemical events in real-time within living cells represents perhaps the most transformative advantage of FRET biosensors. This capability reveals dynamic biological processes that are entirely inaccessible to conventional endpoint assays, providing unprecedented insights into the temporal organization and spatial coordination of signaling networks.
Technical Implementation of Live-Cell FRET Imaging
Real-time live-cell imaging with FRET biosensors can be implemented through multiple detection modalities, each with distinct advantages for specific experimental requirements. Ratiometric imaging measures the emission ratio of acceptor to donor fluorescence, providing a robust readout that is relatively straightforward to implement on standard fluorescence microscopes and corrects for many common artifacts [23] [24]. Fluorescence Lifetime Imaging Microscopy (FLIM) measures the reduction in donor fluorescence lifetime due to FRET, offering superior quantification that is independent of fluorophore concentration and excitation intensity, particularly advantageous in complex samples and deep tissues [26] [24]. For high-throughput applications, plate reader-based FRET detection enables screening of multiple conditions simultaneously, as demonstrated in drug efficacy studies [25] [26]. The development of dark FRET acceptors has further advanced live-cell imaging by enabling multiplexed monitoring of multiple targets without spectral cross-talk, as exemplified by the Multiplexed Dark FRET (MDF) platform [30].
Representative Experimental Protocols
Protocol: Monitoring Kinase Inhibition in Live Cells
This protocol outlines the procedure for evaluating kinase inhibitor efficacy using FRET biosensors in live cells, based on methodologies successfully employed in cancer research [25]:
Biosensor Expression: Introduce the FRET biosensor (e.g., Pickles biosensor for BCR-ABL) into target cells via transfection, electroporation, or viral transduction. Validate expression and proper localization via fluorescence microscopy after 24-48 hours.
Experimental Setup: Seed biosensor-expressing cells into appropriate imaging chambers (e.g., 96-well plates for high-throughput screening or glass-bottom dishes for microscopy) and allow to adhere overnight.
* inhibitor Treatment*: Apply serial dilutions of kinase inhibitors (e.g., imatinib, nilotinib for BCR-ABL) to cells, including DMSO-only controls. For temporal monitoring, add inhibitors directly during image acquisition.
Image Acquisition: Acquire time-lapse FRET images using either:
- Ratiometric Method: Capture donor (e.g., CFP/excitation 430-450 nm, emission 460-500 nm) and acceptor (e.g., YFP/excitation 500 nm, emission 520-550 nm) channels simultaneously or sequentially at 2-5 minute intervals.
- FLIM Method: Measure donor fluorescence lifetime using time-domain or frequency-domain FLIM systems at regular intervals.
Data Analysis:
- For ratiometric data: Calculate FRET ratio (acceptor emission/donor emission) for each time point and normalize to baseline.
- For FLIM data: Analyze fluorescence lifetime decay curves and calculate FRET efficiency from donor lifetime reduction.
- Generate dose-response curves from steady-state measurements to determine IC₅₀ values.
Protocol: Real-Time Visualization of STAT Activation
This protocol describes the procedure for monitoring STAT activation dynamics using genetically encoded biosensors, based on the STATeLight platform [26]:
Biosensor Design and Expression: Fuse full-length or truncated STAT (e.g., STAT5A) C-terminally to selected FRET pair (e.g., mNeonGreen and mScarlet-I) via optimized linkers. Transfect into target cells (e.g., HEK-Blue IL-2 cells for STAT5 studies).
Stimulation and Imaging:
- Serum-starve cells for 4-6 hours prior to imaging to establish baseline activity.
- Stimulate with appropriate cytokine (e.g., IL-2 for STAT5 activation) during continuous image acquisition.
- For FLIM-FRET measurements, collect donor (mNeonGreen) fluorescence lifetime images before and after stimulation.
Image Processing and Analysis:
- Calculate FRET efficiency from donor fluorescence lifetime (τ) using: E = 1 - (τDA/τD), where τDA is donor lifetime with acceptor, and τD is donor lifetime alone.
- Generate kinetic curves of FRET efficiency changes over time.
- Map spatial heterogeneity of STAT activation within individual cells.
The Scientist's Toolkit: Essential Research Reagents and Materials
The successful implementation of FRET biosensor technology requires specific reagents and instrumentation tailored to live-cell imaging applications. The following table catalogues essential components for researchers establishing FRET biosensor capabilities.
Table 3: Essential Research Reagents and Materials for FRET Biosensor Studies
| Category | Specific Examples | Function/Application | Notes |
|---|---|---|---|
| FRET Pairs | CFP/YFP [11], mNeonGreen/mScarlet-I [26], mNg/ShadowY [30] | Donor-acceptor combinations for energy transfer | Dark acceptors (ShadowY, ShadowR) reduce spectral bleed-through [30] |
| Expression Systems | Lentiviral vectors, Plasmid transfection reagents, Stable cell lines | Biosensor delivery into target cells | Viral methods offer higher efficiency for primary cells [25] |
| Imaging Equipment | Fluorescence microscopes with FRET capabilities, FLIM systems, Plate readers with lifetime detection | Signal detection and quantification | FLIM provides more quantitative measurements [26] [24] |
| Cell Culture | Appropriate cell lines (e.g., HEK293T, primary cells), Imaging-optimized media, Chambered coverslips | Maintenance and imaging of biosensor-expressing cells | Low-fluorescence media reduces background [26] |
| Biosensor Constructs | Pickles (BCR-ABL) [25], STATeLights [26], AKAR (PKA) [24], TORCAR (mTOR) [24] | Target-specific activity monitoring | Available from academic collaborators or Addgene |
Genetically encoded FRET biosensors provide an unparalleled toolkit for investigating biochemical processes in living systems, offering significant advantages over conventional techniques in sensitivity, specificity, and temporal resolution. Their ability to detect molecular events at physiologically relevant concentrations with high specificity, while enabling real-time monitoring in live cells, has already transformed our understanding of dynamic cellular processes. Continued refinement of biosensor design—including the development of dark acceptors, expanded color palettes, and improved targeting strategies—promises to further enhance these capabilities. As these technologies become increasingly accessible to the broader research community, they are poised to accelerate both fundamental biological discovery and translational drug development efforts across a spectrum of human diseases.
From Design to Discovery: Advanced Methodologies and Cutting-Edge Applications
Genetically encoded biosensors based on Förster Resonance Energy Transfer (FRET) have revolutionized our ability to monitor cellular processes in living systems with high spatiotemporal resolution. These sophisticated molecular tools typically consist of a sensing domain flanked by two fluorescent proteins (FPs) that form a FRET pair. When the sensing domain interacts with its target analyte or undergoes a conformational change due to enzymatic activity, it alters the distance or orientation between the FPs, resulting in a measurable change in FRET efficiency [23] [31]. For nearly two decades, the cornerstone of FRET biosensor design has relied on the cyan-yellow FP pair (CFP/YFP), which offers reasonable spectral overlap for energy transfer but presents significant limitations for complex experimental applications [32].
The CFP-YFP pair occupies a substantial portion of the visible spectrum, creating challenges for multiplexing with other fluorescent probes. Furthermore, this traditional FRET pair suffers from issues of cross-excitation and spectral bleed-through, where the acceptor (YFP) can be directly excited by wavelengths intended for the donor (CFP), and donor emission can contaminate the acceptor detection channel [32]. Compounding these problems, biological systems exhibit significant autofluorescence in the blue-green spectral range when excited by ultraviolet or blue light, reducing the signal-to-background ratio and limiting detection sensitivity [33]. These constraints have driven the biosensor community to develop a new generation of red-shifted FRET biosensors that operate at longer wavelengths, offering improved spectral characteristics, reduced biological autofluorescence, and enhanced capabilities for multiplexed imaging of multiple biochemical events simultaneously [33].
Fundamental Advantages of Red-Shifted FRET Biosensors
Enhanced Optical Properties for Live-Cell Imaging
The migration of FRET biosensors toward longer wavelengths represents a significant technological advancement with multiple demonstrable benefits. Biological tissues and cells exhibit markedly reduced autofluorescence in the red and near-infrared regions of the spectrum compared to the blue-green range. This inherent property translates directly to improved signal-to-background ratios, enabling detection of weaker signals and smaller FRET changes that might be obscured by background noise in conventional CFP-YFP systems [33]. Additionally, longer wavelength light experiences reduced scattering in biological samples and is less phototoxic to living cells, allowing for prolonged imaging sessions without compromising cellular viability or function [33].
From a practical perspective, red-shifted biosensors offer expanded multiplexing capabilities by freeing up the blue-green spectral regions for other probes. Researchers can now simultaneously monitor multiple signaling pathways by combining red FRET biosensors with green-emitting indicators or synthetic dyes that would normally spectrally overlap with CFP-YFP pairs [32]. This capability is particularly valuable for studying complex signaling networks where understanding the temporal relationships between different biochemical events is crucial. The development of these advanced tools has been facilitated by both the engineering of improved red fluorescent proteins and innovative approaches to FRET pair design [31].
Key Performance Metrics of Red-Shifted FRET Pairs
Table 1: Comparison of representative red-shifted FRET pairs for biosensor development
| FRET Pair (Donor/Acceptor) | Excitation Max (nm) | Emission Max (nm) | FRET Efficiency | Dynamic Range | Key Advantages |
|---|---|---|---|---|---|
| OFP/MFP (mCyRFP1/mMaroon1) | 549 / 586 | 569 / 662 | High | Nearly doubled vs GFP/RFP | Large Stokes shift, improved S/B ratio [33] |
| GFP/RFP | 484 / 555 | 510 / 584 | Moderate | Reference | Better than CFP/YFP [33] |
| miRFP670/miRFP720 | 592 / 637 | 670 / 720 | High | Demonstrated | NIR range, minimal autofluorescence [34] |
| ChemoG5SiR (eGFP-SiR) | 488 / 652 | 510 / 670 | 95.8% | Unprecedented | Chemogenetic, near-quantitative FRET [35] |
Engineering Strategies for Red-Shifted FRET Biosensors
Protein Engineering and Directed Evolution
The development of high-performance red-shifted FRET biosensors has relied heavily on advanced protein engineering strategies. Initial efforts focused on improving the photophysical properties of existing red fluorescent proteins, which traditionally suffered from poor brightness, slow maturation, and tendency to form aggregates [31]. Through systematic directed evolution, researchers addressed these limitations by creating monomeric variants with enhanced quantum yields, improved photostability, and reduced oligomerization tendencies [33] [31].
A landmark achievement in this field came from the development of the OFP/MFP pair (mCyRFP1/mMaroon1), which was specifically optimized for fluorescence lifetime imaging and FRET applications. This pair exhibits exceptional spectral characteristics with well-separated excitation and emission peaks, minimizing cross-talk between channels [33]. The engineering process involved numerous rounds of mutagenesis and screening to improve critical parameters such as maturation efficiency at 37°C, extinction coefficients, and photostability under prolonged illumination. The resulting mMaroon1 acceptor possesses a large Stokes shift and emission maximum at 662 nm, making it particularly suitable for combination with orange fluorescent donors [33].
Chemogenetic FRET Pairs: A Hybrid Approach
A revolutionary approach to FRET biosensor design emerged recently with the development of chemogenetic FRET pairs that combine fluorescent proteins with synthetic fluorophores. The groundbreaking ChemoG5 system exemplifies this strategy, employing an engineered interface between enhanced GFP (eGFP) and a HaloTag7 (HT7) self-labeling protein covalently linked to silicon rhodamine (SiR) [35].
Table 2: The ChemoX palette of chemogenetic FRET pairs
| FRET Construct | Fluorescent Protein | Synthetic Fluorophore | FRET Efficiency | Key Applications |
|---|---|---|---|---|
| ChemoB | eBFP2 | SiR | ≥94% | Blue-shifted multiplexing |
| ChemoC | mCerulean3 | SiR | ≥94% | Cyan replacement |
| ChemoG5 | eGFP | SiR | 95.8% | High-efficiency reference |
| ChemoY | Venus | SiR | ≥94% | Yellow-shifted applications |
| ChemoR | mScarlet | SiR | 91.3% | Fully red-shifted pair |
The engineering of ChemoG5 involved introducing specific interface mutations (eGFP: A206K and T225R; HT7: E143R, E147R and L271E) that stabilize the interaction between the FP and labeled HaloTag, resulting in near-quantitative FRET efficiency of 95.8% [35]. This remarkable efficiency stems from the extremely close proximity (15.2 Å) between the eGFP chromophore and the synthetic fluorophore, as confirmed by X-ray crystallography [35]. The chemogenetic approach provides unparalleled spectral flexibility, as the HaloTag can be labeled with different rhodamine fluorophores to shift the acceptor emission wavelength from 556 nm (with JF525) to 686 nm (with JF669) while maintaining ≥94% FRET efficiency [35].
Near-Infrared FRET Biosensors
Pushing further into the infrared spectrum, researchers have developed near-infrared (NIR) FRET biosensors that utilize specialized fluorescent proteins such as miRFP670 and miRFP720. These pairs operate in the spectral range of 670-720 nm, where tissue autofluorescence is minimal and light penetration is maximal [34]. The NIR FRET Rac1 biosensor exemplifies this technology, incorporating an innovative auto-inhibitory motif with a second p-21 binding domain (PBD) containing GTPase-binding deficient mutations to limit FRET-competent interactions exclusively to the presence of active GTP-bound Rac1 [34]. This design specificity is crucial for accurately monitoring the spatiotemporal dynamics of small GTPase activation in living cells with minimal background signal.
Experimental Implementation and Validation
Protocol: Development and Testing of a Red-Shifted FRET Biosensor
The following protocol outlines the key steps for developing and validating a red-shifted FRET biosensor, based on methodologies successfully employed for the OFP/MFP SERCA2a biosensor and NIR FRET Rac1 biosensor [34] [33]:
Molecular Cloning and Vector Design: Subclone the gene of interest into an appropriate mammalian expression vector, flanked by selected donor and acceptor FPs at strategic positions (N-terminus, C-terminus, or internal loops). For the SERCA2a biosensor, the donor (OFP) was inserted into an internal loop while the acceptor (MFP) was placed at the N-terminus to maximize conformational-dependent FRET changes [33].
Cell Culture and Stable Line Generation: Maintain HEK293 cells in phenol red-free DMEM supplemented with 2 mM GlutaMAX and 10% fetal bovine serum at 37°C with 5% CO2. Generate stable clones expressing the biosensor using appropriate selection antibiotics, with empty plasmid controls for background subtraction [34] [33].
Transfection Optimization: For transient transfections, use polyethylenimine (PEI) as a cost-effective transfection reagent. In a 12-well plate format, transfect with 400 ng of biosensor plasmid in 100 μL OptiMEM solution, with empty plasmid control DNA added to a total of 2 μg as needed [34].
Spectral Scanning and FRET Verification: Acquire fluorescence emission spectra using a microplate reader with monochromators (e.g., CLARIOstar Plus). Set excitation to 591-52 nm and collect emission from 640-15 to 802-15 nm with appropriate gain settings. Perform blank correction using spectra from cells transfected with empty plasmid [34].
Functional Validation: Test biosensor response to known activators and inhibitors. For the NIR Rac1 biosensor, this included co-transfection with activator protein fragments (TrioD1SH3) and negative regulators (GDI), as well as testing of constitutively active (Q61L) and dominant negative (T17N) mutants [34].
High-Throughput Screening Setup: For HTS applications, dispense cells into 1536-well flat, black-bottom polypropylene plates at a density of 10^6 cells/mL using an automated liquid dispenser. Acquire fluorescence lifetime data using a high-throughput microplate reader capable of measuring nanosecond decays [33].
Figure 1: Development workflow for red-shifted FRET biosensors, from initial design to functional application
The Scientist's Toolkit: Essential Reagents and Instruments
Table 3: Key research reagent solutions for red-shifted FRET biosensor development
| Category | Specific Examples | Function/Application |
|---|---|---|
| Fluorescent Proteins | mMaroon1, mCyRFP1, miRFP670, miRFP720 | FRET acceptors in red-shifted pairs |
| Expression Systems | PEI transfection reagent, 12-well TC-treated plates | Biosensor delivery and cell culture |
| Detection Instruments | CLARIOstar Plus with red-extended PMT, Fluorescence lifetime plate readers | Spectral scanning and high-throughput FRET detection |
| Reference Standards | Constitutively active mutants (Q61L), Dominant negative mutants (T17N) | Biosensor validation and normalization |
| Modulator Compounds | TrioD1SH3 (activator), GDI (inhibitor) | Functional testing of biosensor response |
Applications in Multiplexed Imaging and Drug Discovery
Multiparameter Imaging of Signaling Networks
The expanded color palette provided by red-shifted FRET biosensors has enabled unprecedented capabilities for multiparameter imaging of complex signaling networks in live cells. Researchers can now simultaneously monitor multiple biochemical events by combining spectrally distinct biosensors that operate in different wavelength ranges. One successful strategy utilizes spatial separation of spectrally identical biosensors targeted to different subcellular compartments, allowing parallel monitoring of the same biochemical activity in distinct locations [32]. For example, simultaneous imaging of plasma membrane-targeted and nuclear-localized cAMP reporters has revealed compartmentalized signaling dynamics that would be impossible to detect with single biosensors [32].
Advanced approaches employ multiple FRET pairs with distinct spectral signatures to visualize different signaling activities simultaneously. The combination of green-red FRET pairs with blue-yellow pairs enables dual-parameter rationetric imaging, though this requires careful optimization to minimize spectral cross-talk [32]. Alternatively, fluorescence lifetime imaging (FRET-FLIM) provides a powerful solution for multiplexing, as it primarily requires isolation of the donor fluorescence, leaving most of the spectrum available for other probes [32]. This approach has been successfully used to monitor caspase-3 activity alongside intracellular calcium levels using LSSmOrange-mKate2 and CFP-YFP FRET pairs [31].
High-Throughput Screening Applications
Red-shifted FRET biosensors have demonstrated particular utility in high-throughput screening (HTS) platforms for drug discovery. The OFP/MFP SERCA2a biosensor, when coupled with a high-throughput fluorescence lifetime microplate reader, enabled high-precision nanosecond-resolved fluorescence decay measurements from microliter sample volumes, with an entire 1536-well plate read in less than three minutes [33]. This configuration achieved a remarkable 30-fold improvement in detection precision compared to conventional intensity-based assays, allowing identification of subtle structural modulators that would be missed by traditional screening approaches [33].
The advantages of red-shifted biosensors in HTS include reduced compound autofluorescence (as most drug-like molecules fluoresce in the blue-green range), minimized light scattering, and decreased cellular autofluorescence [33]. These factors collectively contribute to significantly improved signal-to-background ratios and better assay quality metrics, ultimately increasing the success rate of identifying true hits in large compound libraries. The implementation of these biosensors in live-cell formats provides the additional advantage of ensuring that identified compounds can penetrate cellular membranes and function under physiological conditions [33].
Figure 2: Application landscape for red-shifted FRET biosensors spanning multiplexed imaging, high-throughput screening, and drug discovery
Future Perspectives and Concluding Remarks
The development of red-shifted FRET biosensors represents a significant milestone in the ongoing evolution of genetically encoded biosensors. The field continues to advance through several promising avenues. Further expansion into the near-infrared spectrum remains an active area of research, with ongoing efforts to develop even longer wavelength FRET pairs that would enable deeper tissue imaging and further reduced background [34]. The chemogenetic approach exemplified by the ChemoX palette offers unprecedented flexibility, and future iterations will likely expand this concept to incorporate additional synthetic fluorophores with optimized photophysical properties [35].
Another exciting frontier involves the development of biosensors with expanded dynamic ranges through computational protein design and machine learning approaches. As demonstrated by the chemogenetic FRET pairs, strategic interface engineering can dramatically improve FRET efficiency and dynamic range, suggesting that similar principles could be applied to more traditional FP-based biosensors [35]. Additionally, the integration of red-shifted biosensors with advanced imaging modalities such as super-resolution microscopy and light-sheet imaging will open new possibilities for visualizing biochemical activities with unprecedented spatial and temporal resolution.
In conclusion, the expanding color palette of red-shifted FRET biosensors has transformed our ability to study complex biological systems in their native contexts. These tools provide enhanced performance characteristics, reduced interference from biological autofluorescence, and unprecedented capabilities for multiparameter imaging. As these technologies continue to mature and become more widely adopted, they will undoubtedly yield new insights into cellular signaling networks and accelerate the discovery of novel therapeutic interventions. The ongoing collaboration between protein engineers, cell biologists, and instrumentation developers ensures that the future of FRET biosensor technology will remain bright—across the entire visible spectrum and beyond.
Cell fate decisions, such as proliferation, differentiation, and survival, are regulated by intricate signaling networks where Extracellular signal-Regulated Kinase (ERK) and Protein Kinase B (AKT) function as central nodes [36]. Understanding the dynamic coordination between these pathways requires tools capable of capturing their spatiotemporal activity in live cells. Genetically encoded fluorescent biosensors have revolutionized cell biology research by enabling real-time monitoring of molecular activities with exceptional spatial and temporal resolution [37]. These biosensors provide critical insights into signaling behaviors that static methods like immunoblotting cannot capture, including oscillatory signaling, propagating waves, and pulsatile activities that govern cellular decision-making processes [37].
Among various designs, Förster Resonance Energy Transfer (FRET)-based biosensors have emerged as particularly powerful tools for quantifying kinase activity [37]. These biosensors typically consist of two fluorescent proteins (FPs) flanking a sensing module that responds to kinase-mediated phosphorylation by inducing a conformational change, altering FRET efficiency between the FPs [36]. The development of increasingly sophisticated FRET biosensors has enabled researchers to decode the complex dynamics of ERK and AKT signaling, revealing how their temporal patterns and spatial compartmentalization contribute to signaling specificity and cellular outcomes [38] [39].
Core Biosensor Designs and Mechanisms
Fundamental FRET Biosensor Architecture
FRET-based kinase biosensors operate on the principle of non-radiative energy transfer between a donor fluorophore and an acceptor fluorophore when they are in close proximity (typically 1-10 nm) [37]. The core architecture of an intramolecular FRET biosensor for kinases typically includes:
- Fluorescent protein pair: A donor FP and acceptor FP with spectral properties suitable for FRET
- Substrate domain: A specific amino acid sequence recognized by the target kinase
- Phospho-recognition domain: A binding domain that recognizes the phosphorylated substrate
- Flexible linkers: Sequences that connect these elements and provide conformational flexibility
Upon phosphorylation by the target kinase, the phospho-recognition domain binds to the phosphorylated substrate, inducing an intramolecular interaction that brings the two FPs into closer proximity, thereby increasing FRET efficiency [36]. This change in FRET can be quantified through ratiometric imaging, providing a quantitative readout of kinase activity [40].
Evolution of ERK Biosensors (EKAR)
The ERK Kinase Activity Reporter (EKAR) family has undergone significant evolution since its initial inception. Early versions like EKAR-EV contained a CFP variant (ECFP) tethered to a YFP variant (YPet) via a flexible linker housing an ERK substrate domain and a WW phospho-amino acid binding domain [36]. Subsequent improvements included:
- Brighter and more pH-stable CFP and YFP variants [36]
- Optimization of linker length to modulate dynamic range and sensitivity [36]
- Repositioning of fluorescent proteins to enhance FRET efficiency [38]
- Reduction of non-specific phosphorylation through amino acid substitutions to eliminate CDK-mediated signals during G2/M phase [36]
Recent iterations include EKAREN4 and EKAREN5, which feature different dynamic ranges optimized for specific applications. EKAREN4 has a larger dynamic range and higher saturation point, while EKAREN5 offers greater sensitivity for detecting low levels of ERK activity [36]. The development of EKAR4 represented another significant advancement, created by switching the positions of the fluorescent proteins in EKAR-EV to yield a 74.5% increase in the emission ratio response in PC-12 cells [38].
AKT Biosensor Development
For monitoring AKT activity, researchers have developed several FRET-based biosensors. The Eevee-iAkt represents a highly specific AKT biosensor based on the Eevee backbone [41]. This biosensor was rigorously validated using inhibitors targeting kinases upstream and downstream of AKT, confirming its specificity for monitoring Akt activity in living cells [41]. The development of targeted versions enabled visualization of Akt activity in different subcellular compartments, including raft and non-raft regions of the plasma membrane, mitochondria, and nucleus [41].
Another significant advancement came with the ExRai-AKTAR2, a cpGFP-based AKT biosensor noted for its high performance [36]. However, its spectral properties overlap with CFP/YFP FRET pairs, creating challenges for multiplexing with conventional EKAR biosensors [36].
Table 1: Evolution of Key Kinase Biosensors
| Biosensor | Target | FRET Pair/Signal | Key Features | Applications |
|---|---|---|---|---|
| EKAR-EV | ERK | CFP/YFP | Original EKAR design | General ERK activity monitoring |
| EKAR2G/EKAR3 | ERK | CFP/YFP | Improved linkers | Enhanced temporal resolution |
| EKAREN4/5 | ERK | CFP/YFP | Reduced CDK1 cross-talk; different dynamic ranges | EKAREN4: high activity; EKAREN5: low activity |
| EKAR4 | ERK | CFP/YFP | Flipped FP positions | 74.5% increased response; spatial targeting |
| REKAR67/76 | ERK | miRFP670nano3/miRFP720 | Red-shifted emission (670-720 nm) | Multiplexing with CFP/YFP biosensors |
| Eevee-iAkt | AKT | FRET-based | High specificity; targetable | Subcellular AKT activity |
| ExRai-AKTAR2 | AKT | cpGFP-based | High performance | AKT activity, but limited multiplexing capability |
Recent Technological Advances
Spectral Separation: Red-Shifted ERK Biosensors
A significant limitation in multiplexed biosensing has been the spectral overlap between biosensors. To address this, researchers recently developed two novel red-FRET ERK biosensors, REKAR67 and REKAR76, which operate in the 670-720 nm range using the fluorescent proteins miRFP670nano3 and miRFP720 [36]. These biosensors were created as red-shifted versions of EKAREN4 by replacing the traditional CFP and YFP fluorophores with far-red alternatives [36].
The two REKAR variants differ in fluorophore positioning, which significantly impacts their performance characteristics:
- REKAR67: Features miRFP670nano3 at the N-terminus followed by miRFP720, displays a higher dynamic range but greater signal variance [36]
- REKAR76: Features miRFP720 at the N-terminus followed by miRFP670nano3, shows reduced signal variance with a more moderate dynamic range [36]
In comparative studies, both REKAR biosensors demonstrated high consistency with existing CFP/YFP biosensors in reporting ERK activity following EGF stimulation and inhibition by EGFR and MEK inhibitors [36]. Their spectral compatibility with CFP/YFP FRET and cpGFP-based biosensors enables multiplexed imaging of multiple signaling pathways simultaneously [36].
Spatial Targeting of Biosensors
Subcellular compartmentalization of kinase signaling adds another layer of regulatory complexity. To address this, researchers have developed targeted versions of biosensors localized to specific subcellular regions. For example, EKAR4 has been successfully targeted to:
- Cytoplasm: Using a nuclear export signal (NES) [38]
- Nucleus: Using a nuclear localization sequence (NLS) [38]
- Plasma membrane: Using the hypervariable region of KRas including a polylysine and CAAX box sequence [38]
These targeted biosensors have revealed striking differences in ERK activity dynamics at different subcellular locations. While EGF induces transient ERK activity in the cytosol and nucleus, it stimulates sustained ERK activity at the plasma membrane [38]. This sustained plasma membrane ERK activity involves Rap1, a noncanonical activator, and controls cell morphology and EGF-induced membrane protrusion dynamics [38].
Diagram 1: FRET Biosensor Working Mechanism
Engineering Approaches and Optimization Strategies
The development of highly sensitive FRET biosensors has been accelerated through systematic optimization approaches. Key advancements include:
Backbone Optimization: Researchers have developed an optimized backbone for intramolecular FRET biosensors featuring an optimized pair of fluorescent proteins and long flexible linkers ranging from 116 to 244 amino acids in length [40]. This design reduces basal FRET signals and increases the gain of FRET biosensors by rendering them completely "distance-dependent" rather than "orientation-dependent" [40].
High-Throughput Screening (FRET-Seq): Recent innovations integrate FRET sorting with next-generation sequencing (FRET-Seq) to identify sensitive biosensors from large-scale libraries directly in mammalian cells [42]. This approach utilizes self-activating FRET (saFRET) biosensors fused to an active kinase domain, allowing for high-throughput screening of biosensor variants with enhanced performance [42].
FP Pair Optimization: Systematic evaluation of FP pairs has identified optimal combinations for distance-dependent FRET biosensors. The ECFP/YPet and Turquoise-GL/YPet pairs have demonstrated the largest gain in FRET/CFP ratio, with dimerization-prone FP pairs proving particularly suitable for distance-dependent FRET biosensors [40].
Table 2: Performance Comparison of ERK Biosensors
| Biosensor | Dynamic Range | Spectral Range | Spatial Resolution | Key Advantages | Limitations |
|---|---|---|---|---|---|
| EKAR-EV | Moderate (~30-40%) | CFP/YFP (450-530 nm) | Cytosolic | Established protocol | Spectral overlap |
| EKAR2G/EKAR3 | Improved (~50%) | CFP/YFP (450-530 nm) | Cytosolic | Better signal-to-noise | Still limited multiplexing |
| EKAR4 | High (~75%) | CFP/YFP (450-530 nm) | Targetable | Large dynamic range | Spectral overlap |
| EKAREN4 | High | CFP/YFP (450-530 nm) | Cytosolic | Reduced CDK1 cross-talk | Spectral overlap |
| EKAREN5 | High sensitivity for low activity | CFP/YFP (450-530 nm) | Cytosolic | Detects low ERK activity | Spectral overlap |
| REKAR67 | High (higher than REKAR76) | 670-720 nm | Cytosolic | Multiplexing capability | Higher signal variance |
| REKAR76 | Moderate (lower variance) | 670-720 nm | Cytosolic | More consistent signals | Lower dynamic range |
Experimental Methodology
Biosensor Implementation and Live-Cell Imaging
Implementing FRET biosensors for kinase signaling studies requires careful experimental design and execution. A typical workflow includes:
Biosensor Delivery: REKAR and EKAR biosensors are typically delivered via lentiviral transduction to ensure stable expression [36] [43]. For example, in developing REKAR biosensors, researchers transduced MCF-10A cells with pLenti-REKAR viral particles produced by transfecting HEK 293T cells with FuGENE HD transfection reagent [43]. Transduction is performed in polybrene-treated media with centrifugation at 100 × g for 30 minutes, followed by flow cytometry sorting to isolate biosensor-positive cells [43].
Cell Culture and Preparation: Human mammary epithelial cells (MCF-10A) are cultured in growth medium supplemented with appropriate factors [36]. For imaging with far-red biosensors like REKAR, cells require supplementation with 25 μM biliverdin, a chromophore precursor necessary for miRFP670nano3 and miRFP720 maturation [43]. Cells are plated on collagen-coated glass-bottom imaging plates at least 48 hours before experiments and transferred to imaging medium 16 hours prior to imaging [43].
Live-Cell Imaging: Imaging experiments are performed using advanced microscopy systems such as a Nikon Eclipse Ti2 inverted microscope equipped with a Teledyne Photometrix Kinetix sCMOS camera and a 20X/0.75 NA Plan Apo objective [43]. Cells are maintained at 37°C with 5% CO2 during imaging. For temporal dynamics studies, images are typically captured every 6 minutes to balance temporal resolution with phototoxicity concerns [43].
Stimulation and Inhibition: Experiments typically include stimulation with Epidermal Growth Factor (EGF) to activate ERK signaling, and inhibition using compounds such as:
- PD-0325901: A MEK inhibitor that blocks ERK activation [36]
- Gefitinib (ZD1839): An EGFR inhibitor that upstream of ERK signaling [36]
Diagram 2: Experimental Workflow for Biosensor Imaging
Image Acquisition and FRET Quantification
FRET imaging typically employs ratiometric quantification, where cells are excited at the donor excitation wavelength and emissions are collected from both donor and acceptor channels [40]. The FRET/CFP ratio is used to represent the level of FRET efficiency, which correlates with kinase activity [40]. For REKAR biosensors using miRFP670nano3 and miRFP720, appropriate filter sets are selected to match their excitation and emission spectra in the 670-720 nm range [36].
Data processing is often performed using customized algorithms in MATLAB, with previously described methods available through GitHub repositories such as https://github.com/Albeck-Lab/REKAR [43]. The signal is typically normalized and expressed as a percentage increase over basal signal, allowing for quantitative comparisons across experiments and conditions [38].
Multiplexed Biosensor Applications
Strategies for Multiplexed Imaging
A primary challenge in multiplexed biosensor imaging is spectral overlap between fluorescent proteins. Innovative strategies have been developed to address this limitation:
Spectral Separation: By carefully selecting FPs with minimal spectral overlap, multiple biosensors can be expressed and imaged simultaneously in the same cells [37]. The development of red-shifted biosensors like REKAR67 and REKAR76 enables spectral compatibility with CFP/YFP FRET and cpGFP-based biosensors, allowing dual-color imaging of multiple signaling pathways [36].
Linear Unmixing: For higher levels of multiplexing, FPs with significant spectral overlaps can be distinguished using spectral imaging followed by linear unmixing [37]. This method assumes that the total measured fluorescence at each wavelength is a linear combination of signals from all fluorophores present. Using known emission spectra of individual fluorophores, linear unmixing can determine the relative contribution of each fluorophore, enabling simultaneous imaging of up to five or six different fluorophores [37].
Temporal Differentiation: Photochromic or reversibly switching FPs can be exploited to distinguish signals based on temporal responses to specific illumination patterns rather than solely spectral properties [37].
Spatial Segregation: Targeting biosensors to specific subcellular compartments or using cell barcoding techniques provides another dimension for distinguishing multiple signals [37].
Integrated ERK and AKT Signaling Studies
Multiplexed imaging of ERK and AKT pathways is particularly valuable since these pathways often cooperate in regulating cell fate decisions. For example, measuring ERK and AKT activity together has provided insights into their combined roles in promoting cell proliferation or differentiation [43]. Studies have revealed how these pathways interact during processes such as collective cell migration, tissue repair, and response to growth factors [37].
The development of spectrally distinct biosensors now enables researchers to simultaneously monitor ERK and AKT activities with subcellular resolution in the same cell, revealing how their spatial and temporal coordination determines cellular outcomes [36]. This capability is particularly important for understanding complex processes such as cancer progression, where multiple signaling pathways are frequently dysregulated.
Research Reagent Solutions
Table 3: Essential Research Reagents for Biosensor Experiments
| Reagent/Category | Specific Examples | Function/Application | Experimental Notes |
|---|---|---|---|
| Cell Lines | MCF-10A (human mammary epithelial) | Model system for signaling studies | Clone 5E preferred for consistency [36] |
| Biosensor Plasmids | pLV x EF1a_EKAREN4-ires-puro, pLenti-REKAR67/76 | Source of biosensor constructs | Available from Addgene [36] |
| Transfection Reagents | FuGENE HD Transfection Reagent | Lentiviral production | HD-1000 [36] |
| Molecular Cloning | Gibson Assembly Master Mix | Biosensor construction and modification | NEB M5510A [36] |
| Extracellular Matrix | Collagen I, Rat tail | Cell plating substrate | Promotes proper cell adhesion [36] |
| Signaling Modulators | EGF (Epidermal Growth Factor) | ERK pathway activation | Peprotech Cat#AF-100-15 [36] |
| Inhibitors | PD-0325901 (MEK inhibitor), Gefitinib (EGFR inhibitor) | Pathway inhibition | Validate biosensor specificity [36] |
| Imaging Plates | Glass bottom plates, #1.5H high performance | High-resolution live imaging | Cellvis P96-1.5H-N [36] |
| Chromophore Supplements | Biliverdin | Essential for miRFP670nano3/miRFP720 | 25 μM concentration [43] |
| Software | NIS-Elements AR, MATLAB, Harmony | Image acquisition and analysis | Custom analysis code available [36] [43] |
The continued evolution of FRET-based biosensors for ERK and AKT signaling has dramatically enhanced our ability to monitor kinase dynamics in live cells with high spatiotemporal resolution. Recent developments, particularly the creation of red-shifted biosensors like REKAR67 and REKAR76, address the critical challenge of spectral overlap that has limited multiplexed imaging of signaling networks [36]. These advancements, combined with improved biosensor sensitivity through optimized backbone designs and high-throughput screening approaches, provide powerful tools for deciphering the complex dynamics of cell fate decisions [40] [42].
Future directions in biosensor development will likely focus on several key areas. First, expanding the color palette further into the near-infrared spectrum will enable even greater multiplexing capabilities [37]. Second, combining biosensors with cutting-edge techniques such as optogenetics will allow not only observation but also precise manipulation of signaling pathways to establish causal relationships [39]. Third, integrating biosensor data with artificial intelligence-driven analysis holds great potential for uncovering complex patterns in cellular decision-making processes [37]. Finally, continued innovation in biosensor design and implementation will deepen our understanding of molecular networks in cells, with significant implications for both fundamental biology and therapeutic development [37].
As these technologies mature, they will increasingly enable researchers to move beyond studying signaling pathways in isolation toward a more integrated understanding of how networks of interacting molecules coordinate to determine cellular behaviors in health and disease.
Förster Resonance Energy Transfer (FRET) biosensors have emerged as powerful tools in clinical diagnostics, enabling the detection of pathogens with high sensitivity, specificity, and rapidity. FRET is a phenomenon involving non-radiative energy transfer through dipole-dipole coupling between two fluorophores—a donor and an acceptor—when they are in close proximity (typically 1-10 nm) [4]. The efficiency of this energy transfer is highly sensitive to the distance and orientation between the fluorophores, making FRET an excellent mechanism for reporting molecular interactions and conformational changes [4]. In pathogen detection, this technology has been successfully applied to identify various targets, including the SARS-CoV-2 virus responsible for the COVID-19 pandemic. The design of these biosensors often incorporates genetically encoded elements or nanomaterial components, allowing for precise detection of viral proteins or nucleic acids within a framework that aligns with broader biosensor development research [35]. The integration of FRET biosensors into diagnostic platforms represents a significant advancement over traditional methods, offering the potential for point-of-care testing that is both rapid and reliable, which is crucial for timely disease management and outbreak control.
Fundamental Principles of FRET
Theoretical Foundation
The efficiency (E) of FRET is quantitatively described by Förster's theory, which establishes that E is inversely proportional to the sixth power of the distance (r) between the donor and acceptor fluorophores, and directly proportional to the Förster radius (R₀), as defined by the equation E = 1 / [1 + (r/R₀)⁶] [4]. The R₀ value represents the distance at which 50% energy transfer occurs and is specific to each FRET pair. It depends on several photophysical parameters: the quantum yield of the donor (QD), the spectral overlap integral (J) between the donor's emission spectrum and the acceptor's excitation spectrum, the relative orientation of the donor and acceptor dipoles (κ²), and the refractive index of the medium (n) [4]. This strong distance dependence makes FRET exceptionally useful for monitoring molecular interactions and conformational changes at the nanoscale.
Design Considerations for FRET Biosensors
The design of effective FRET biosensors requires careful selection of donor-acceptor pairs with significant spectral overlap to facilitate efficient energy transfer [35]. Common pairs include CFP-YFP for genetically encoded sensors or quantum dots with organic dyes in nanomaterial-based sensors [4]. The biosensor typically consists of a sensing domain that recognizes the specific pathogen biomarker (e.g., antibody, nucleic acid probe) coupled with the FRET pair. When the target biomarker interacts with the sensing domain, it induces a conformational change that alters the distance or orientation between the donor and acceptor, resulting in a measurable change in FRET efficiency [35]. This change is typically quantified by monitoring the emission ratio of the acceptor to donor fluorescence, known as the FRET ratio, which provides a sensitive readout of the target analyte's presence and concentration [4].
FRET-Based Detection of SARS-CoV-2
Spike Protein Detection Using Nanomaterial-Based FRET
A highly sensitive FRET-based immunoassay has been developed for detecting SARS-CoV-2 Spike (S) protein. This method employs carbon quantum dots (CQDs) as donors and gold nanoparticles (AuNPs) as acceptors, both modified with SARS-CoV-2 antibodies [44]. In the absence of the target S protein, the CQDs and AuNPs are in close proximity, enabling FRET to occur and quenching the CQD fluorescence. When the S protein is present, it binds to the antibodies on both nanoparticles, increasing the distance between donors and acceptors, thereby reducing FRET efficiency and restoring CQD fluorescence in a concentration-dependent manner—a mechanism known as the "turn-off-on" mode [44]. This assay demonstrates exceptional performance with a detection limit of 0.05 ng/mL and a broad linear range of 0.1–100 ng/mL. The entire detection process requires only 12 minutes, significantly faster than conventional RT-PCR methods [44]. Furthermore, the platform has been validated for compatibility with human serum and effectiveness against SARS-CoV-2 variants (B.1.1.529 and B.1.617.2), highlighting its clinical relevance [44].
Protease Activity Detection via FRET Peptide Substrates
An alternative FRET-based approach targets SARS-CoV-2 protease activity as a diagnostic biomarker. This method utilizes specific fluorogenic peptide substrates containing a fluorophore (FITC) and a quencher (dabcyl) at opposite termini [45]. In the intact peptide, the proximity of the quencher to the fluorophore suppresses fluorescence. When SARS-CoV-2 proteases cleave the peptide at specific recognition sites, the fluorophore and quencher separate, resulting in a measurable increase in fluorescence intensity [45]. Through screening a library of 115 dipeptide sequences, researchers identified optimal substrates that SARS-CoV-2 proteases selectively cleave. This assay achieves a detection limit of 9.7 ± 3 pfu/mL and shows no cross-reactivity with MERS-CoV, confirming high specificity [45]. The significant fluorescence changes observed with patient samples indicate strong potential for clinical diagnosis using this protease-based FRET assay.
Nucleic Acid Detection with Quantum Dot FRET
Quantum dots (QDs) have been utilized in FRET-based nucleic acid sensors for detecting SARS-CoV-2 genetic material. One innovative platform employs primer-passivated CdTe QDs functionalized with thiol-labeled primers specific to conserved regions of the SARS-CoV-2 genome [46]. In this design, the QDs serve as donors, and a dye-labeled complementary sequence acts as the acceptor. When the target viral RNA is present, it facilitates sandwich hybridization, bringing the QD and dye into close proximity and enabling FRET to occur [46]. This system achieves a detection limit of 1.71 × 10⁻⁹ mol/L and provides results within 15 minutes, showing strong correlation with RT-PCR outcomes when tested on actual patient samples [46]. The platform demonstrates how QD-FRET technology can provide rapid, sensitive, and specific detection of viral nucleic acids, making it suitable for point-of-care diagnostic applications.
Table 1: Performance Comparison of FRET-Based Methods for SARS-CoV-2 Detection
| Detection Target | FRET Pair | Limit of Detection | Linear Range | Assay Time | Reference |
|---|---|---|---|---|---|
| Spike Protein | CQDs / AuNPs | 0.05 ng/mL | 0.1–100 ng/mL | 12 min | [44] |
| Protease Activity | FITC / Dabcyl | 9.7 ± 3 pfu/mL | Not specified | Not specified | [45] |
| Viral RNA | CdTe QDs / Dye | 1.71 × 10⁻⁹ mol/L | Not specified | 15 min | [46] |
Advanced FRET Biosensor Design and Engineering
Chemogenetic FRET Pairs with Enhanced Dynamic Range
Recent breakthroughs in FRET biosensor design have led to the development of chemogenetic FRET pairs with near-quantitative FRET efficiencies. These innovative systems combine fluorescent proteins (FPs) with synthetic fluorophores bound to self-labeling proteins such as HaloTag [35]. For example, engineering an interface between enhanced GFP (eGFP) and a silicon rhodamine (SiR)-labeled HaloTag (HT7) has yielded a FRET pair with efficiency exceeding 95% [35]. This remarkable efficiency was achieved through rational mutagenesis of interface residues (eGFP: A206K and T225R; HT7: E143R, E147R, and L271E) to stabilize the interaction between the FP and labeled HaloTag [35]. The chemogenetic design offers exceptional spectral tunability, allowing researchers to select different FP components (e.g., eBFP2, mCerulean3, Venus, mScarlet) and various rhodamine fluorophores (e.g., JF525, TMR, JF669) to create FRET pairs with desired spectral properties [35]. This versatility enables multiplexed detection schemes and optimization for specific diagnostic applications while maintaining high dynamic ranges and sensitivity.
Near-Infrared FRET Biosensors for Improved Imaging
The development of near-infrared (NIR) FRET biosensors represents a significant advancement for biological detection and imaging. NIR FRET pairs, such as miRFP670 and miRFP720, offer distinct advantages including reduced autofluorescence, deeper tissue penetration, and minimal phototoxicity [34]. These properties make NIR FRET biosensors particularly valuable for live-cell imaging and potential in vivo applications. For instance, a NIR FRET biosensor for Rac1 GTPase activity has been successfully validated, demonstrating expected FRET changes in response to mutations and co-expression of modifying proteins [34]. The incorporation of such NIR pairs into pathogen detection systems could enhance sensitivity in complex biological samples and enable more sophisticated diagnostic applications with improved signal-to-noise ratios.
Table 2: Advanced FRET Technologies and Their Applications
| Technology Platform | Key Components | FRET Efficiency | Advantages | Potential Diagnostic Applications |
|---|---|---|---|---|
| Chemogenetic FRET | FP + Rhodamine-labeled HaloTag | ≥94% | High dynamic range, spectral tunability | Multiplexed pathogen detection, live-cell imaging |
| Near-Infrared FRET | miRFP670 / miRFP720 | Not specified | Reduced autofluorescence, deeper tissue penetration | In vivo sensing, complex sample analysis |
| Quantum Dot FRET | CdTe QDs / Organic Dyes | Not specified | High brightness, photostability | Rapid nucleic acid detection, point-of-care diagnostics |
Experimental Protocols
Protocol: FRET-Based SARS-CoV-2 Spike Protein Detection
Materials and Reagents
- Carbon quantum dots (CQDs), 10 mg/mL
- Gold nanoparticles (AuNPs), negatively charged (﹣AuNPs) or positively charged (﹢AuNPs)
- Anti-SARS-CoV-2 spike RBD neutralizing antibody
- SARS-CoV-2 trimer S protein (target analyte)
- EDC (N-(3-Dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride)
- Sulfo-NHS (N-hydroxysulfosuccinimide)
- MES buffer (0.1 mol/L, pH = 5.5)
- PBS buffer (50 mmol/L, pH = 7.4)
- BSA (bovine serum albumin, ≥98%)
- Ultrafiltration centrifuge tubes (30 kDa MWCO)
Synthesis of Positively Charged AuNPs (﹢AuNPs)
- Add 500 μL of 216 mmol/L cysteamine solution to 200 mL of 0.01% (w/V) chloroauric acid solution.
- Stir for 20 minutes at 400 rpm at room temperature.
- Quickly add 10 μL of 10 mmol/L sodium borohydride solution.
- Increase stirring speed to 500 rpm and react vigorously for 15 minutes.
- Store the resulting wine-red ﹢AuNPs at 4°C [44].
Preparation of Antibody-Conjugated CQDs (CQDs-Ab)
- Dilute 100 μL of 10 mg/mL CQDs solution to 5 mL with MES buffer (pH = 5.5).
- Add 0.8 mg EDC and 2.2 mg Sulfo-NHS, then react for 15 minutes.
- Add 1.5 mL PBS (pH = 13) to adjust the solution pH to 7.6.
- Add 200 μL of 0.2 mg/mL anti-SARS-CoV-2 spike RBD neutralizing antibodies and react for 2 hours.
- Add 200 μL of 2 mg/mL BSA solution and incubate for 30 minutes to block non-specific sites.
- Concentrate the mixture using 30 kDa ultrafiltration centrifuge tubes (RCF 3743 g, 10 minutes, 4°C).
- Recover the conjugated CQDs-Ab in 2 mL solution [44].
Detection Assay Procedure
- Mix CQDs-Ab with ﹢AuNPs in appropriate proportions.
- Add sample containing SARS-CoV-2 S protein.
- Incubate for 12 minutes at room temperature.
- Measure fluorescence emission using a fluorescence spectrometer with excitation at 360 nm.
- Quantify S protein concentration based on the increase in fluorescence intensity at 450 nm [44].
Protocol: FRET-Based Protease Detection for SARS-CoV-2
Materials
- FRET peptide substrate with FITC and dabcyl
- SARS-CoV-2 virus sample
- Vero E6 cells for virus propagation
- DMEM with 10% FBS
- HEPES buffer
Assay Procedure
- Design FRET peptide substrates with fluorophore (FITC) and quencher (dabcyl) at opposite termini.
- Incubate peptide substrate with SARS-CoV-2 sample containing viral proteases.
- Monitor fluorescence intensity increase over time (excitation 485-495 nm, emission 515-525 nm).
- Quantify viral load based on the rate of fluorescence increase or endpoint measurement [45].
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Research Reagent Solutions for FRET-Based Pathogen Detection
| Reagent/Material | Function/Application | Examples/Specifications |
|---|---|---|
| Carbon Quantum Dots (CQDs) | FRET donor | Strong fluorescence, modifiable surface, high stability [44] |
| Gold Nanoparticles (AuNPs) | FRET acceptor | Tunable surface charge, strong quenching ability [44] |
| HaloTag System | Self-labeling protein platform | Covalent binding to synthetic fluorophores, chemogenetic FRET [35] |
| Rhodamine Fluorophores | Synthetic FRET acceptors | JF525, TMR, SiR; spectral tunability [35] |
| Quantum Dots (QDs) | Nanomaterial FRET donors | CdTe QDs; high brightness, narrow emissions [46] [47] |
| Fluorogenic Peptide Substrates | Protease activity detection | FITC-dabcyl labeled, specific cleavage sequences [45] |
| EDC/Sulfo-NHS | Crosslinking chemistry | Antibody-nanoparticle conjugation [44] |
Schematic Diagrams of FRET Detection Mechanisms
SARS-CoV-2 Spike Protein Detection via Nanoparticle FRET
Nucleic Acid Detection via Quantum Dot FRET
FRET-based biosensors represent a transformative approach in clinical diagnostics for pathogen detection, offering significant advantages in sensitivity, speed, and specificity compared to traditional methods. The applications in SARS-CoV-2 detection—targeting spike proteins, protease activity, and nucleic acids—demonstrate the versatility and effectiveness of this technology. Ongoing advancements in chemogenetic FRET pairs, near-infrared imaging, and nanomaterial integration continue to push the boundaries of what's possible in diagnostic biosensing. As these technologies mature, FRET-based biosensors are poised to play an increasingly important role in global health security, enabling rapid response to emerging infectious diseases and improving patient care through point-of-care testing capabilities.
Förster Resonance Energy Transfer (FRET)-based biosensors represent a powerful class of genetically encoded tools that enable the monitoring of molecular events in living systems with high spatiotemporal resolution. The principle of FRET involves non-radiative energy transfer from an excited donor fluorophore to a nearby acceptor fluorophore when they are within close proximity (typically 1-10 nm), with efficiency inversely proportional to the sixth power of the distance between them [8] [48]. This exquisite distance dependence makes FRET ideal for reporting conformational changes in proteins, protein-protein interactions, and enzymatic activities in live cells and tissues [49] [50].
The phosphatase and tensin homolog (PTEN) is a critical tumor suppressor protein that serves as a central regulator of cellular metabolism, proliferation, and growth by antagonizing the PI3K/AKT/mTOR signaling pathway [51] [52]. PTEN dephosphorylates phosphatidylinositol 3,4,5-trisphosphate (PIP3), thereby inhibiting downstream Akt activation and cellular growth pathways [51]. Despite its fundamental importance in development, synaptic plasticity, and tumor suppression, and its association with numerous human pathologies including cancer, autism spectrum disorder, and epilepsy, methods to directly monitor PTEN activity dynamics in intact biological systems have been historically limited [51] [52]. Traditional approaches relying on indirect biochemical assays or genetic manipulations could not provide the temporal or spatial resolution needed to understand PTEN signaling in its native context.
Recent breakthroughs have addressed this technological gap through the development of conformation-sensitive PTEN biosensors optimized for fluorescence lifetime imaging microscopy (FLIM), particularly in neural systems [51] [53]. This whitepaper examines the design principles, validation methodologies, and applications of these novel biosensors, focusing on their implementation for monitoring PTEN dynamics in the intact brain.
PTEN FRET-FLIM Biosensor Design and Engineering
Structural Basis for PTEN Conformational Sensing
The design of the PTEN biosensor capitalizes on the natural conformational dynamics of the PTEN protein. Structural studies have revealed that PTEN undergoes a transition between a closed, inactive conformation and an open, active state [51] [53]. This conformational change is regulated by phosphorylation of serine residues in the C-terminal region by casein kinase 2 (CK2), which promotes the closed, inactive form [51]. When PTEN transitions to its active state, it adopts a more extended conformation, increasing the distance between its N- and C-termini.
The biosensor engineering strategy involved tagging the N- and C-termini of PTEN with donor and acceptor fluorescent proteins, respectively, creating a molecular ruler that reports conformational changes through modulation of FRET efficiency [51]. The specific construct utilizes monomeric enhanced GFP (mEGFP) as the FRET donor and sREACh, a dark YFP variant, as the acceptor [51]. This particular donor-acceptor combination is optimized for fluorescence lifetime imaging measurements, as changes in GFP lifetime provide a robust, quantitative readout of FRET efficiency that is relatively insensitive to fluorescence fluctuations and sensor expression levels [51].
Table 1: Key Components of the PTEN FRET-FLIM Biosensor
| Component | Description | Function |
|---|---|---|
| PTEN Backbone | Wild-type or mutant PTEN protein (403 amino acids) | Senses conformational changes correlated with activity state |
| Donor Fluorophore | Monomeric Enhanced GFP (mEGFP) | FRET donor; exhibits lifetime changes responsive to energy transfer |
| Acceptor Fluorophore | sREACh (dark YFP variant) | FRET acceptor; quenches donor fluorescence without emitting |
| Linker Regions | Optimized peptide sequences | Connects fluorophores to PTEN; influences dynamic range |
| Point Mutation | R14G substitution | Reduces catalytic activity while maintaining conformational sensitivity |
Biosensor Optimization and Validation
Extensive optimization efforts focused on refining linker regions between PTEN and the fluorescent proteins to maximize the dynamic range of the biosensor [51]. The optimal configuration demonstrated high basal FRET levels (reflected by low fluorescence lifetime of 2.20 ± 0.003 ns) compared to donor-only tagged PTEN (2.69 ± 0.001 ns), indicating a predominantly closed conformation under resting conditions [51].
A critical innovation in the biosensor development was the identification of a point mutation (R14G) that preserves 95% of the conformational sensitivity while reducing catalytic activity to approximately 5% of wild-type PTEN [52]. This engineered biosensor acts as a "minimally perturbing" reporter that can be expressed without significantly interfering with endogenous PTEN signaling or cellular function, as demonstrated by the normal density of dendritic spines in mouse somatosensory cortical neurons expressing the sensor [52].
The biosensor was validated through multiple pharmacological and genetic approaches. Treatment with tetrabromobenzotriazole (TBB), a CK2 inhibitor that promotes PTEN activation, induced a sustained increase in fluorescence lifetime (0.23 ± 0.003 ns), reflecting decreased FRET due to PTEN adopting an open conformation [51]. Conversely, epidermal growth factor (EGF), which negatively regulates PTEN activity, decreased fluorescence lifetime (from 2.23 ± 0.003 ns to 2.17 ± 0.003 ns), indicating increased FRET consistent with a more closed conformation [51]. The reversibility of these conformational dynamics was confirmed through sequential application of TBB followed by washout and EGF treatment [51].
Diagram 1: PTEN Conformational Dynamics and FRET Biosensing Principle. CK2 phosphorylation promotes a closed PTEN conformation with high FRET between terminal fluorophores. Activation leads to an open conformation with increased distance between fluorophores and reduced FRET efficiency, detected as increased fluorescence lifetime.
Experimental Methodology and Technical Implementation
Fluorescence Lifetime Imaging Microscopy (FLIM)
The PTEN biosensor is optimized for two-photon fluorescence lifetime imaging microscopy (2pFLIM), a technique that provides several advantages for in vivo brain imaging. Unlike intensity-based FRET measurements, FLIM quantifies the average time a fluorophore remains in the excited state before emitting a photon, a parameter that is independent of fluorophore concentration, excitation light intensity, and photon pathlength [51] [49]. This makes FLIM particularly suitable for quantitative measurements in complex tissue environments where these factors can vary.
In the implemented methodology, the fluorescence lifetime of the mEGFP donor is measured, with decreased lifetime indicating increased FRET efficiency (due to the additional energy transfer pathway) and thus a more closed PTEN conformation [51]. The typical lifetime values observed range from approximately 2.1-2.2 ns for the closed conformation to 2.5-2.6 ns for the open conformation, with differences as small as 0.03-0.05 ns being statistically significant in well-controlled experiments [51] [52].
Table 2: Representative FLIM Lifetime Values Under Various Conditions
| Experimental Condition | FLIM Lifetime (ns) | Interpretation |
|---|---|---|
| Donor-only PTEN | 2.69 ± 0.001 | Baseline (no FRET) |
| Biosensor - Basal | 2.20 ± 0.003 | High FRET (Closed conformation) |
| Biosensor - 4A mutant | 2.53 ± 0.002 | Low FRET (Open conformation) |
| TBB treatment | Increase of 0.23 ± 0.003 | Increased PTEN activity |
| EGF treatment | Decrease of 0.06 ± 0.003 | Decreased PTEN activity |
| Neuronal cell body | 2.20 ± 0.004 | Higher PTEN activity |
| Dendritic region | 2.11 ± 0.009 | Lower PTEN activity |
In Vivo Imaging Protocols
For in vivo imaging in the mouse brain, the PTEN biosensor is typically delivered via adeno-associated virus (AAV) vectors, with expression driven by cell-type-specific promoters such as synapsin for neuronal expression [54]. Surgical procedures involve cranial window implantation to provide optical access to the cortex, followed by a recovery period before imaging sessions [51].
Two-photon imaging is performed through the cranial window using lasers tuned to the appropriate excitation wavelength for mEGFP (approximately 920 nm). Fluorescence lifetime data are collected using time-correlated single photon counting (TCSPC) systems, which provide the precise timing information needed for lifetime calculations [51]. Imaging sessions can be repeated over days or weeks to track longitudinal changes in PTEN activity.
For experiments involving sensory manipulation, mice are typically anesthetized and presented with controlled stimuli, while simultaneous calcium imaging can be performed using red-shifted indicators in combination with the red-shifted R-PTEN biosensor variant [51] [52].
Diagram 2: Experimental Workflow for In Vivo PTEN FLIM Imaging. Key steps from biosensor delivery through surgical preparation, data acquisition, and analysis enable quantitative monitoring of PTEN activity dynamics in the intact brain.
Research Applications and Key Findings
Cell-Type and Subcellular Compartment-Specific PTEN Dynamics
The implementation of the PTEN FRET-FLIM biosensor has revealed unprecedented spatial and temporal heterogeneity in PTEN activity within the intact brain. A notable finding concerns the subcellular compartmentalization of PTEN signaling, with neuronal cell bodies exhibiting significantly higher PTEN activity (2.20 ± 0.004 ns) compared to dendritic regions (2.11 ± 0.009 ns) [52]. This spatial gradient suggests distinct regulatory mechanisms and functional roles for PTEN in different neuronal compartments.
The development of a red-shifted sensor variant (R-PTEN) enabled cell-type-specific monitoring of PTEN dynamics through simultaneous imaging with GFP-based calcium indicators [51] [52]. This multispectral approach revealed that sensory experience drives divergent PTEN signaling pathways in excitatory versus inhibitory neurons. Specifically, sensory deprivation led to decreased PTEN activity in excitatory neurons (approximately 0.04 ns decrease) while increasing PTEN activity in inhibitory neurons (approximately 0.15 ns increase) [52]. This "molecular seesaw" pattern provides mechanistic insight into how balanced network activity is maintained and how it may become disrupted in neurodevelopmental disorders.
Developmental and Pathological PTEN Regulation
Applications across model systems have demonstrated the versatility of the biosensor for investigating PTEN regulation in various biological contexts. In C. elegans, PTEN activity was shown to gradually increase throughout development, with fluorescence lifetime values progressing from 2.51 ns in L1 larvae to 2.64 ns in adults [52]. This developmental regulation aligns with PTEN's established role in controlling cell growth and metabolic processes.
The biosensor also enables investigation of disease-associated PTEN mutations, many of which cause structural destabilization and misfolding [51]. By monitoring how pathogenic mutations affect PTEN conformation in living cells, researchers can dissect the molecular mechanisms underlying PTEN-related pathologies and potentially identify approaches to rescue function of destabilized mutants.
Research Reagent Solutions
The following table summarizes key reagents developed for PTEN FLIM imaging, with plasmids available through Addgene [54]:
Table 3: Essential Research Reagents for PTEN FLIM Studies
| Reagent Name | Description | Primary Application |
|---|---|---|
| CMV_G-PTEN | Green PTEN biosensor under CMV promoter | In vitro and cell culture studies |
| CMV_R-PTEN | Red PTEN biosensor under CMV promoter | Dual-color imaging with GFP-based sensors |
| CMVG-PTEN4A | Constitutively active mutant (4A) | Positive control for open conformation |
| CAG_G-PTEN | Green biosensor under CAG promoter | Strong, consistent expression in neurons |
| AAV-pSyn_R-PTEN | AAV with red biosensor under synapsin promoter | In vivo neuronal expression |
| AAV-FLEX-CAG_G-PTEN | Cre-dependent AAV for green biosensor | Cell-type-specific expression in transgenic lines |
The development of conformation-sensitive PTEN biosensors for FLIM imaging represents a significant advancement in our ability to study signaling dynamics in intact biological systems. The technical approach described herein enables quantitative monitoring of PTEN activity with subcellular resolution in living cells, intact tissues, and whole organisms, providing unprecedented insight into the spatial and temporal regulation of this critical tumor suppressor.
Future developments in this field will likely focus on expanding the color palette of biosensors to enable simultaneous monitoring of multiple signaling pathways, further improving signal-to-noise ratios, and developing even less perturbing reporters that can be knock-in expressed at endogenous loci. Combined with ongoing advancements in microscopy technology, particularly in deep-tissue imaging approaches, these tools will continue to illuminate the dynamic landscape of intracellular signaling in health and disease.
The integration of these biosensors with other modalities, including electrophysiology, behavioral monitoring, and transcriptomic analysis, will provide increasingly comprehensive understanding of how PTEN signaling contributes to neural circuit function and dysfunction. As these tools become more widely adopted, they will accelerate both basic research into PTEN biology and translational efforts to target PTEN-related pathways for therapeutic benefit.
Recent advances in drug discovery have established biosensors as indispensable tools, particularly valued for their precision, sensitivity, and real-time monitoring capabilities. Genetically encoded fluorescent biosensors have revolutionized the study of cell signaling and metabolism, allowing live-cell measurements with high spatiotemporal resolution that capture dynamic cellular processes previously inaccessible with endpoint assays [55]. These biosensors generally consist of a sensing unit that responds to an analyte or enzyme activity by undergoing conformational change, and a reporting unit that transduces this change into a measurable fluorescent signal [55]. In high-throughput compound screening, this capability enables researchers to monitor compound effects on specific molecular targets within physiological cellular environments, providing more physiologically relevant data compared to traditional biochemical assays.
The integration of biosensors in high-throughput screening (HTS) has enabled breakthrough discoveries in targeted cancer therapies and other therapeutic areas. Biosensors have revealed complex signaling dynamics within cells, including oscillatory signaling, molecular waves, and pulsatile activities that regulate critical cellular functions and disease processes [37]. These dynamic behaviors occur across multiple scales, from subcellular regions to whole cell populations, and are essential to a wide range of biological functions including cell proliferation, migration, and stress responses [37]. The ability to capture these temporal and spatial patterns during compound screening provides invaluable insights for drug development that static assays cannot deliver.
Biosensor Types and Mechanisms for Screening Applications
Primary Biosensor Designs for Drug Screening
Biosensors used in high-throughput drug discovery employ several distinct mechanisms to report molecular events, each with advantages for specific screening applications:
FRET-Based Biosensors: These sensors rely on Förster Resonance Energy Transfer between two fluorophores. A conformational change in the sensing unit alters the distance or orientation between donor and acceptor fluorophores, changing FRET efficiency [37]. This design is particularly valuable for monitoring protein-protein interactions, kinase activities, and second messenger dynamics [56] [55]. FRET changes can be quantified through multiple methods including fluorescence lifetime imaging (FLIM), donor-acceptor emission ratio measurements, or acceptor photobleaching [57] [37].
Single-Fluorophore Intensity Biosensors: These utilize circularly permuted fluorescent proteins (cpFPs) that change fluorescence intensity upon conformational changes induced by target molecule binding [37]. Examples include the GCaMP series for calcium sensing and GRAB sensors for neuropeptide detection [37]. Their simplicity makes them suitable for higher throughput applications, though they may be more susceptible to false positives from compound autofluorescence or effects on expression levels.
Translocation Biosensors: These sensors undergo subcellular redistribution in response to molecular events, detectable as changes in fluorescence localization [37]. Kinase Translocation Reporters (KTRs) combine nuclear localization signals (NLS), nuclear export signals (NES), and kinase-specific phosphorylation sites to create phosphorylation-dependent nucleocytoplasmic shuttling [37]. The PH domain of AKT kinase fused to fluorescent proteins translocates to the plasma membrane upon PI3K activation through PIP3 binding [37].
Spectral Shift Biosensors: These exhibit altered excitation or emission profiles in response to molecular events, enabling rationetric measurements that are less sensitive to variations in biosensor concentration or excitation intensity [37].
Table 1: Biosensor Types and Their Applications in Compound Screening
| Biosensor Type | Detection Method | Primary Applications in Screening | Advantages for HTS |
|---|---|---|---|
| FRET-Based | FRET efficiency changes via emission ratios or FLIM | Protein-protein interactions, kinase activity, second messengers | Quantitative, internally controlled, sensitive to molecular proximity |
| Single-Fluorophore Intensity | Fluorescence intensity changes | Ion concentrations, metabolite levels, small molecule detection | Simplified optics, compatible with standard plate readers |
| Translocation | Subcellular redistribution | Kinase activity, lipid second messengers, signaling pathway activation | Visual context, high content information |
| Spectral Shift | Excitation/emission ratio changes | Metabolite levels, ion concentrations, pH | Ratiometric quantification reduces artifacts |
Advanced Biosensor Technologies
Recent engineering efforts have produced biosensors with enhanced capabilities for drug discovery:
Near-Infrared FRET Biosensors: Utilizing fluorophores like miRFP670 and miRFP720, these sensors enable spectral resolution from biosensors in the blue/green/yellow range while reducing cellular autofluorescence [34]. This allows multiplexing with existing biosensors and improves signal-to-noise ratios in complex cellular environments.
Chemigenetic Biosensors: These combine self-labeling protein tags (HaloTag, SNAP-tag) with synthetic fluorophores, offering narrower emission spectra than fluorescent proteins and facilitating multiplexed imaging [37]. The exogenous fluorophores provide enhanced photostability and brightness compared to genetic fluorophores.
BRET and Bioluminescence Sensors: Bioluminescence Resonance Energy Transfer (BRET) systems use luciferase-generated light as the donor source, eliminating the need for excitation illumination and reducing autofluorescence [56]. NanoBRET and NanoBiT technologies have been successfully applied to monitor protein-protein interactions and compound effects in live cells [56].
Biosensor Applications in High-Throughput Compound Screening
Successful Drug Discovery Applications
Biosensors have enabled several significant advances in targeted cancer therapy and drug discovery:
The identification of Celastrol as a novel YAP-TEAD inhibitor was achieved through NanoBiT-based screening, demonstrating how biosensor platforms can reveal novel mechanisms of natural compounds [56]. In another example, TR-FRET assays successfully identified Ro-31-8220 as a SMAD4R361H/SMAD3 interaction inducer, illustrating how biosensors can uncover new therapeutic strategies for targeting specific cancer mutations [56].
The development of LATS biosensors revealed VEGFR as an upstream regulator of the Hippo signaling pathway, connecting two important signaling networks with implications for cancer therapy [56]. Similarly, NanoBRET assays detecting RAF dimerization have provided insights into the mechanisms of RAF inhibitors, important targeted therapies for melanoma and other cancers [56]. HiBiT systems monitoring protein degradation dynamics have enabled real-time tracking of targeted protein degradation compounds, an emerging therapeutic modality [56].
Biosensors for small GTPases have been particularly valuable, as these proteins regulate critical processes in cancer including cell growth, movement, and cytoskeletal dynamics [34]. The NIR FRET Rac1 biosensor has enabled validation of Rac1 mutations and screening for compounds that modulate GTPase activity, with spectral scanning demonstrating clear FRET changes in response to activators and inhibitors [34].
Quantitative Biosensor Performance in Screening
Biosensors provide quantitative readouts of compound effects on specific molecular targets within their physiological cellular context:
Table 2: Quantitative Biosensor Performance in Compound Screening Applications
| Biosensor Target | Biosensor Technology | Key Performance Metrics | Screening Applications |
|---|---|---|---|
| cAMP Signaling | Epac-based FRET-FLIM | Lifetime change from 3.92±0.08 ns (control) to 2.93±0.11 ns (activated); EFRET,open = 25.3%±3.2% [57] | GPCR drug screening, phosphodiesterase inhibitors |
| Rac1 GTPase | NIR FRET (miRFPs) | Significant FRET increase with Q61L mutation (p<0.01); decreased with effector binding mutations [34] | Anti-metastatic compounds, cytoskeletal modulators |
| Kinase Activities | FRET-based / KTR | Specific phosphorylation-dependent conformational changes or localization [37] | Kinase inhibitor screening, pathway analysis |
| Protein Degradation | HiBiT / NanoBiT | Real-time monitoring of protein stability and degradation kinetics [56] | PROTACs, molecular glues, protein homeostasis modulators |
| YAP-TEAD Interaction | NanoBiT | Identification of novel inhibitors like Celastrol [56] | Cancer therapeutics targeting Hippo pathway |
Experimental Protocols for Biosensor-Based Screening
FRET Biosensor Validation and Screening Protocol
The following methodology outlines a standardized approach for implementing FRET biosensors in high-throughput compound screening:
Cell Preparation and Transfection:
- Plate HEK293 or other relevant cell lines in clear, tissue-culture treated microplates (e.g., 12-well Costar plates) at optimized densities [34].
- Transfect cells with biosensor plasmids (e.g., 400 ng for 6-well format, adjusted for plate size) using transfection reagents such as PEI [34].
- Include appropriate controls: empty vector, constitutive active mutants (e.g., Rac1 Q61L), dominant negative mutants (e.g., Rac1 T17N), and effector binding mutants (e.g., Rac1 T35S-Y40C) [34].
- For multiplexed experiments, co-transfect with modifying proteins (e.g., activator fragments like TrioD1SH3 or inhibitors like GDI) to validate biosensor response [34].
Instrumentation and Data Acquisition:
- Utilize plate readers with spectral scanning capabilities such as the CLARIOstar Plus with red-extended PMT [34].
- Configure instrument settings: excitation at 591-52 nm, emission scanning from 640-15 to 802-15 nm, gain at 2600, focal height at 4.8 mm, and 60 flashes per read with 0.5-second settling time [34].
- Perform measurements in clear L15 media with 10% FBS at room temperature [34].
- For FLIM measurements, use parallelized confocal systems with 64 beamlets for improved temporal resolution (~0.5 frames per second) with low power per beamlet (~1-2 μW) to minimize photodamage [57].
Data Analysis and Validation:
- Collect spectral data and blank-correct using spectra from cells transfected with empty plasmid controls [34].
- Scale data using reference wavelengths (e.g., RFU at 676 nm as 100%) and analyze FRET changes at target emissions (e.g., 721 nm for NIR FRET) [34].
- For FLIM data, use phasor plots to determine parameters for multi-exponential fitting and calculate FRET efficiency changes [57].
- Validate biosensor performance by confirming expected FRET responses to known activators/inhibitors and mutant constructs before proceeding to compound screening [34].
Multiplexed Biosensor Screening Workflow
Simultaneous monitoring of multiple targets provides comprehensive insights into compound effects and pathway crosstalk:
Spectral Multiplexing Approach:
- Select biosensors with minimal spectral overlap, preferably combining different color variants (e.g., yellow/green with red FPs) [37].
- For higher multiplexing capacity, employ spectral imaging with linear unmixing, enabling simultaneous imaging of up to five or six different fluorophores [37].
- Utilize chemigenetic biosensors with synthetic fluorophores for narrower emission spectra and reduced crosstalk [37].
- Near-infrared FRET biosensors (e.g., miRFP670/miRFP720 pairs) can be combined with visible-range biosensors for simultaneous monitoring of multiple signaling pathways [34].
Temporal and Spatial Multiplexing:
- Implement sequential activation and readout of biosensors using photochromic or reversibly switching fluorescent proteins [37].
- Utilize spatial segregation by targeting biosensors to distinct subcellular compartments or employing cell barcoding techniques [37].
- Combine biosensors with different response kinetics to distinguish immediate compound effects from secondary responses.
Biosensor-Based Screening Workflow
Research Reagent Solutions for Biosensor Screening
Successful implementation of biosensor screening platforms requires specific reagents and instrumentation:
Table 3: Essential Research Reagents and Instruments for Biosensor Screening
| Category | Specific Examples | Function in Screening | Key Features |
|---|---|---|---|
| Cell Lines | HEK293, HeLa | Provide cellular context for biosensor expression | High transfection efficiency, consistent growth |
| Biosensor Plasmids | mTurq2-Epac1-tddVenus [57], NIR FRET Rac1 [34] | Report on specific molecular targets | Optimized dynamic range, spectral properties |
| Transfection Reagents | PEI (Polyethylenimine) [34] | Introduce biosensor DNA into cells | Cost-effective, high efficiency [34] |
| Microplates | Clear, TC-treated (e.g., Costar) [34] | Cell housing during screening | Optimal optical properties, cell attachment |
| Detection Instruments | CLARIOstar Plus [34], Confocal FLIM [57] | Measure biosensor signals | Spectral scanning, sensitivity, temporal resolution |
| Control Reagents | Forskolin/IBMX [57], GTPase mutants [34] | Validate biosensor performance | Known effects on biosensor response |
Technical Considerations and Implementation Challenges
While biosensors offer powerful capabilities for drug discovery, several technical challenges must be addressed for successful implementation in high-throughput screening:
Sensitivity and Stability Considerations: Maintaining consistent sensitivity in HTS applications remains challenging due to variations in biosensor expression, cellular autofluorescence, and compound interference [56]. Environmental factors such as pH, temperature, and oxidative stress can affect biosensor performance in complex cellular environments [56]. Strategies to address these challenges include:
- Implementing ratiometric measurements to normalize for expression variability
- Utilizing fluorescence lifetime measurements (FLIM) to provide intensity-independent readouts [57]
- Incorporating control biosensors with fixed FRET efficiencies to monitor environmental effects
- Using near-infrared biosensors to reduce autofluorescence interference [34]
Multiplexing Limitations and Solutions: Spectral overlap between biosensors limits the number of simultaneous measurements possible [37]. Innovative solutions include:
- Expanding the fluorescent protein palette with brighter, more photostable, and spectrally distinct variants [37]
- Employing temporal multiplexing with photochromic or reversibly switching fluorescent proteins [37]
- Implementing spatial segregation by targeting biosensors to distinct subcellular compartments [37]
- Utilizing computational unmixing algorithms to resolve overlapping signals [37]
Biosensor Design Principles
Future Directions and Emerging Technologies
The field of biosensor development for drug discovery continues to evolve with several promising directions:
Integration with Artificial Intelligence: AI-driven analysis of biosensor data holds great potential for uncovering complex cellular decision-making processes and predicting compound efficacy [56] [37]. Machine learning algorithms can extract subtle patterns from multidimensional biosensor data that may not be apparent through conventional analysis.
Advanced Materials and Nanotechnology: Incorporating quantum dots, upconverting nanoparticles, and other advanced nanomaterials may enhance biosensor performance through improved brightness, photostability, and novel sensing mechanisms [56]. These materials could enable new biosensor designs with enhanced sensitivity and multiplexing capabilities.
Expanded Therapeutic Applications: While cancer drug discovery has been a primary application area, biosensors are increasingly being applied to neurological disorders, metabolic diseases, and infectious diseases [55]. The development of biosensors for new target classes will further expand their utility in drug discovery.
In Vivo and Tissue-Level Screening: Advances in biosensor design and imaging technologies are enabling more physiological screening in complex tissue environments and eventually in live organisms [55] [37]. This progression toward more physiologically relevant screening environments promises to improve the translation of screening hits to clinical candidates.
The continued innovation in biosensor technology coupled with advanced screening platforms will deepen our understanding of molecular networks in cells and accelerate the development of novel therapeutics across disease areas.
Enhancing Performance: Strategies for Biosensor Optimization and Calibration
In the design of genetically encoded FRET biosensors, the dynamic range—the signal difference between the active and inactive states—is a paramount performance metric. While the choice of fluorescent proteins and sensing domains often receives primary focus, the linker regions that connect these components are critical, yet sometimes underestimated, determinants of biosensor sensitivity. Linkers are not merely passive spacers; their composition, length, and rigidity directly govern the distance and orientation of the FRET pair, thereby dictating the efficiency of energy transfer and the magnitude of observable signal change. This technical guide examines the role of linkers in optimizing biosensor dynamic range, focusing on the transformative potential of structured linkers like ER/K α-helical peptides, and provides a framework for their rational implementation within broader FRET biosensor design research for drug development and basic science.
The Critical Role of Linkers in FRET Biosensor Performance
Förster Resonance Energy Transfer (FRET) is a distance-dependent phenomenon sensitive to changes in the nanometer scale, typically operating within a range of 1-10 nm [58]. In single-chain intramolecular FRET biosensors, the conformational change induced by a target analyte alters the proximity of a donor and acceptor fluorophore. The linker domains that tether the sensing and reporting units must therefore permit this structural rearrangement while minimizing unfavorable interactions that could constrain the biosensor's movement or stabilize unproductive conformations.
A key challenge in biosensor engineering is the basal FRET signal in the inactive state. If the donor and acceptor are inherently too close in the "off" state, the dynamic range is severely limited [58]. Early optimization strategies often focused on empirical testing of linker length, but contemporary approaches leverage a deeper understanding of linker biophysics to achieve more predictable and dramatic improvements in performance. The primary goal is to maximize the change in donor-acceptor separation upon biosensor activation, a parameter directly influenced by linker properties.
Figure 1: Conceptual relationship between linker properties and key biosensor performance metrics.
Linker Design Strategies: From Flexible Spacers to Rigid Helices
Flexible Linkers
Traditional biosensor designs often employ long, flexible linkers composed of glycine and serine repeats (e.g., GGS repeats). These linkers provide high degrees of freedom but can result in a high basal FRET due to random coil collapse, which brings the fluorophores into close proximity even in the inactive state. A systematic study by Komatsu et al. demonstrated that flexible linkers ranging from 116 to 244 amino acids could be optimized to reduce basal FRET and increase signal gain by effectively separating the FRET pair in the off-state [40]. The underlying principle is to render FRET efficiency primarily distance-dependent rather than orientation-dependent, which is more difficult to control predictably [40].
Rigid ER/K α-Helical Linkers
A transformative advancement in linker technology is the adoption of ER/K linkers. These peptides consist of alternating repeats of four glutamate (E) and four arginine (R) or lysine (K) residues, which form an extended, stable α-helix with hinge-like properties [58]. The rigidity of this structure prevents the fluorophores from approaching each other in the biosensor's inactive state, thereby minimizing the baseline FRET signal. Upon activation, the defined helical motion facilitates a large, predictable conformational change, leading to a significant increase in FRET efficiency and a greatly expanded dynamic range.
Quantitative Impact of ER/K Linkers: Research on Rac1 FRET and LRET (Lanthanide-based FRET) biosensors has quantified the profound effect of ER/K helix length. For LRET biosensors incorporating a Tb(III) donor and EGFP acceptor, the dynamic range increased with linker length, reaching a remarkable 1100% for a 30 nm ER/K linker [58]. In conventional FRET biosensors with mCerulean and YPet, ER/K linkers also produced significant enhancements, with dynamic ranges up to 125%—a substantial improvement over sensors with unstructured linkers [58].
Table 1: Performance Comparison of Linker Types in Representative Biosensors
| Biosensor Target | Linker Type | FRET/LRET Pair | Reported Dynamic Range | Key Finding | Source |
|---|---|---|---|---|---|
| Rac1 (LRET) | ER/K α-helical (30 nm) | Tb(III) / EGFP | Up to 1100% | Dynamic range increased with ER/K linker length. | [58] |
| Rac1 (FRET) | ER/K α-helical (20 nm) | mCerulean / YPet | Up to 125% | Significant enhancement over unstructured linkers. | [58] |
| PKA, ERK, etc. | Long Flexible (116-244 aa) | ECFP / YPet | Not specified | Reduced basal FRET, enabling rapid biosensor development. | [40] |
| ERK (REKAR67) | Optimized Flexible Linker | miRFP670nano3 / miRFP720 | High (relative to REKAR76) | FP position and linker design impact dynamic range and variance. | [36] |
Experimental Protocols for Linker Optimization
Protocol: Evaluating ER/K Linker Length in LRET Biosensors
This protocol is adapted from studies that achieved a 1100% dynamic range in Rac1 biosensors [58].
Vector Construction:
- Design a biosensor with the domain order: acceptor FP (e.g., EGFP) -> Rac1 binding domain (PBD) -> ER/K linker -> eDHFR -> full-length Rac1.
- Create constructs with varying ER/K linker lengths (e.g., corresponding to 10 nm, 20 nm, and 30 nm helical lengths). The ER/K sequence is typically composed of (EEEEXXXX)n repeats, where X is R or K, and
ndetermines the length.
Cell Transfection and Lysate Preparation:
- Transfect an appropriate cell line (e.g., HEK293) with the biosensor constructs.
- After 24-48 hours, lyse the cells to obtain crude lysates containing the expressed biosensor.
Time-Gated Luminescence (TGL) Measurement:
- Incubate the lysates with a TMP-conjugated Tb(III) complex to label the eDHFR domain.
- In a 96-well plate, use a plate reader capable of time-gated luminescence detection.
- Excite the Tb(III) donor with a pulsed UV light source (∼270-280 nm).
- Introduce a delay (e.g., 50-100 μs) after the excitation pulse to allow short-lived background fluorescence to decay.
- Measure the Tb(III) donor emission at ∼490 nm and the sensitized EGFP acceptor emission at ∼510 nm.
- Calculate the LRET ratio as (Acceptor Emission / Donor Emission).
Data Analysis:
- Stimulate Rac1 activity in lysates (e.g., using GTPγS) and measure the LRET ratio change.
- Quantify the dynamic range as: % Dynamic Range = [(LRETActive - LRETInactive) / LRET_Inactive] × 100.
- Compare the dynamic ranges across the different ER/K linker lengths to identify the optimal construct.
Protocol: Directed Evolution for Comprehensive Biosensor Optimization
Directed evolution is a powerful, high-throughput method to optimize all biosensor components, including linkers, without requiring prior structural knowledge [59] [60].
Library Generation:
- Create a large library (>65,000 variants) of your biosensor gene using error-prone PCR or by introducing degeneracy in the linker-encoding sequences.
- Clone the resulting library into a mammalian expression vector.
High-Throughput Screening via FACS:
- Transduce the plasmid library into mammalian cells (e.g., HEK293 or a relevant lymphocyte line for immune sensors).
- Stimulate the cells under conditions that activate the target (e.g., Zap70 in T-cells) and a separate set under basal conditions.
- Use Fluorescence-Activated Cell Sorting (FACS) to isolate two populations: i) cells with a high FRET ratio (activated state) and ii) cells with a low FRET ratio (basal state).
- The FRET ratio is typically measured as the emission of the acceptor (e.g., YFP) divided by the emission of the donor (e.g., CFP) upon donor excitation.
Sequence Analysis:
- Extract genomic DNA from the sorted cell populations.
- Amplify the biosensor gene sequences and subject them to next-generation sequencing (NGS).
- Analyze the NGS data to identify enriched mutations and linker sequences in the high-FRET population compared to the low-FRET population and the original library.
Validation:
- Clone the identified optimized variants and characterize them in live cells using microscopy or plate readers to confirm enhanced dynamic range and specificity.
Figure 2: A generalized workflow for optimizing biosensor linkers and other components using directed evolution and FACS screening [59] [60].
The Scientist's Toolkit: Essential Reagents for Linker Optimization
Table 2: Key Research Reagents and Resources for Biosensor Engineering
| Reagent / Resource | Function in Optimization | Example Use Case | Source / Reference |
|---|---|---|---|
| ER/K Linker Sequences | Provides a rigid, extended α-helical spacer to minimize basal FRET and maximize conformational change. | Enhancing dynamic range in Rac1 FRET/LRET biosensors. | [58] |
| Fluorescent Protein Pairs (e.g., ECFP/YPet, Turquoise-GL/YPet) | Acts as the FRET reporting unit. Optimized pairs are crucial for high FRET efficiency and signal-to-noise. | Used as the optimized pair in a backbone for developing kinase/GTpase biosensors. | [40] |
| Lanthanide Complexes (Tb³⁺-TMP) | Serves as a long-lifetime LRET donor, enabling time-gated detection to eliminate autofluorescence. | Used in combination with ER/K linkers for high-sensitivity plate-reader assays. | [58] |
| Directed Evolution Platform | A systematic method to generate and screen vast numbers of biosensor variants for desired properties. | Optimizing Zap70 biosensor sensitivity and specificity. | [59] |
| Time-Gated Luminescence Plate Reader | Essential instrument for detecting LRET signals by eliminating short-lived background fluorescence. | Quantifying Rac1 activity in cell lysates with high signal-to-background. | [58] |
Integration with Broader Biosensor Design and Future Perspectives
Optimizing linker design is not an isolated endeavor but must be integrated with other aspects of biosensor engineering. The choice of linker can interact with the selected fluorescent proteins; for instance, dimerization-prone FPs like ECFP/YPet may perform well in distance-dependent designs with long flexible linkers [40]. Furthermore, linker optimization complements advances in sensing domains, such as the use of circularly permuted FPs (cpFPs) and specific point mutations to enhance affinity and specificity [60].
The future of linker optimization lies in the development of more sophisticated and modular design strategies. The integration of computational modeling and machine learning with high-throughput experimental data, such as that from directed evolution, will enable more predictive design of linkers [60]. Furthermore, the push for multiplexed imaging in complex tissues is driving the development of biosensors with red-shifted spectra, where the principles of linker optimization remain equally critical [36]. As these tools evolve, rationally designed linkers will continue to be a fundamental element in unlocking the full potential of FRET biosensors to decipher signaling networks in health and disease.
Fluorescent proteins (FPs) have revolutionized biological imaging by enabling non-invasive visualization of cellular processes in real time. Derived primarily from marine organisms such as the jellyfish Aequorea victoria and coral species Discosoma striata, these proteins form a chromophore within their β-barrel structure that absorbs and emits light at specific wavelengths [61] [62]. The engineering of FPs with enhanced properties—including brightness, photostability, and pH resistance—has become crucial for their application in genetically encoded biosensors, particularly those based on Förster Resonance Energy Transfer (FRET). FRET biosensors allow researchers to monitor dynamic molecular interactions, such as protein-protein interactions, enzyme activities, and metabolite concentrations, within living cells and organisms [63] [64]. The performance of these biosensors directly depends on the optical characteristics of the FPs employed, driving continuous innovation in FP engineering to push the boundaries of sensitivity, duration, and precision in live-cell imaging.
Fundamental Properties of Fluorescent Proteins
Structural Basis of Fluorescence
The core structure of all FPs is a highly stable β-barrel conformation, composed of 11 β-strands forming a hollow barrel that encloses the chromophore [61]. This chromophore is formed spontaneously through chemical modification of specific amino acid residues within the protein chain (typically at positions 65-67 in GFP-like proteins) [62]. The β-barrel structure serves to protect the chromophore from external environmental interference, conferring high thermal and photostability that allows stable luminescence even in complex cellular environments [61]. The specific chemical structure of the chromophore and its immediate amino acid environment determine the spectral properties of the FP, including its excitation and emission wavelengths and sensitivity to environmental factors such as pH.
Key Optical Properties for Biosensing
Three primary optical properties define the utility of FPs in biosensing applications:
- Brightness: Defined as the product of the molar extinction coefficient (ability to absorb photons) and the quantum yield (efficiency of emitting absorbed photons) [61]. Higher brightness enables stronger signals and improved signal-to-noise ratios.
- Photostability: The resistance to photobleaching under sustained illumination, critical for extended imaging sessions [65] [66].
- pH Resistance: The ability to maintain consistent fluorescence across physiological pH variations, essential for reliable performance in diverse cellular compartments [62].
Additional properties including maturation efficiency, monomeric state, and environmental sensitivity also significantly impact FP performance in biosensor applications.
Protein Engineering Strategies
Rational Protein Design
Rational design employs structural knowledge to make targeted mutations that enhance specific FP properties. This approach has successfully improved pH resistance in yellow FPs (YFPs) through mutations such as Q69M (Citrine) that reduce acid sensitivity [62]. Similarly, mutations at critical positions (F223R, L221K, and A206K) where non-polar amino acids are replaced by hydrophilic ones have reduced the tendency for dimerization, preventing artifacts in protein localization studies [62]. Rational design typically requires detailed understanding of the relationships between protein structure and function, leveraging X-ray crystallography data and computational modeling to predict beneficial mutations.
Directed Evolution
Directed evolution mimics natural selection through iterative rounds of mutagenesis and screening to isolate variants with desired traits. This approach does not require prior structural knowledge and can identify beneficial mutations through functional selection. A prominent example is the development of mGold2s and mGold2t, YFPs with up to 25-fold greater photostability than commonly used YFPs, which were identified using a high-throughput pooled single-cell platform that simultaneously screened for both brightness and photostability [65]. Similarly, mRuby3, the brightest and most photostable monomeric red FP characterized to date, was engineered through six rounds of semi-rational evolution that sampled mutations at sites with sequence variability among RFPs [66].
De Novo Protein Design
De novo design represents the most advanced engineering approach, creating entirely novel protein scaffolds with customized properties. While less commonly applied to FPs than other methods, this strategy has potential for developing specialized biosensors with unique characteristics. The review on protein engineering for electrochemical biosensors highlights this as one of three main strategies, though specific examples in FP engineering are less documented in the provided literature [67].
Table 1: Protein Engineering Strategies and Their Applications
| Strategy | Key Features | Examples | Advantages | Limitations |
|---|---|---|---|---|
| Rational Design | Structure-guided targeted mutations | Citrine (Q69M for pH resistance) [62]; A206K for reduced dimerization [62] | Precise; minimal irrelevant mutations | Requires extensive structural knowledge |
| Directed Evolution | Random mutagenesis + high-throughput screening | mGold2s/mGold2t (photostable YFPs) [65]; mRuby3 (bright RFP) [66] | Can discover unexpected solutions; no structural knowledge needed | Resource-intensive screening required |
| De Novo Design | Creating novel protein scaffolds | Limited examples in provided literature [67] | Potential for completely new functions | Theoretically and computationally challenging |
Experimental Protocols for Engineering and Validation
High-Throughput Screening for Photostability and Brightness
The development of mGold2 variants exemplifies a modern approach to FP engineering, utilizing an upgraded version of the SPOTlight single-cell screening platform [65]. The methodology can be summarized as follows:
- Library Construction: Random mutagenesis of the mGold template gene using error-prone PCR or other mutagenesis methods to create a diverse variant library.
- Transformation and Expression: Library transformation into yeast cells for expression, leveraging the eukaryotic host for proper folding and maturation.
- Optical Labeling and Imaging: Individual cells are optically labeled using a strong photoactivation light (4.9 W), reducing labeling time to 10-15 seconds per cell, a significant improvement over previous 45-60 second requirements.
- Parallelized Measurement: Simultaneous measurement of brightness (initial fluorescence intensity) and photostability (fluorescence decay rate under continuous illumination) for millions of cells.
- Cell Sorting: Fluorescence-activated cell sorting (FACS) to isolate cells exhibiting both high brightness and photostability, using mTurquoise2 as a reference FP for normalizing cell-to-cell expression variation.
- Colony Expansion and Validation: Sorted cells are grown into colonies, expanded, and validated through population-level measurements before sequencing top performers.
This platform screened 1,125,438 cells representing 204,987 variants across 7 rounds to identify mGold2s and mGold2t, which contained 14 and 15 mutations relative to mGold, respectively [65].
Characterization of Engineered Fluorescent Proteins
Comprehensive characterization of novel FPs should include both in vitro and cellular assessments:
- Spectroscopic Analysis: Measure excitation and emission spectra using spectrophotometers; determine extinction coefficients and quantum yields using purified proteins [65] [66].
- Photostability Assays: Quantify photobleaching half-lives under defined illumination conditions (e.g., widefield illumination at specific wavelengths and intensities) in live cells and with purified proteins [65].
- pH Sensitivity Testing: Determine pKa values by measuring fluorescence intensity across a pH range, often using buffers with incremental pH changes [62].
- Oligomerization State Assessment: Evaluate monomeric status using techniques such as gel filtration chromatography, critical for ensuring minimal interference with fusion protein function [66].
- Cellular Performance Validation: Test FP fusions with subcellular targeting domains to confirm proper localization and absence of cytotoxic effects [66].
- Functionality in Biosensors: Incorporate superior FPs into established FRET biosensors to validate performance improvements in physiological measurements [65] [66].
Diagram 1: High-throughput FP screening workflow. The process begins with template gene mutagenesis and proceeds through expression, screening, and validation to identify improved variants for biosensor applications. [65]
Quantitative Comparison of Engineered Fluorescent Proteins
Table 2: Photophysical Properties of Representative Engineered Fluorescent Proteins
| Protein | Color | Ex λ (nm) | Em λ (nm) | Brightness* | Photostability | pKa | Key Engineering Features | Reference |
|---|---|---|---|---|---|---|---|---|
| mGold2s | Yellow | ~514 | ~527 | 85 (mClover3 norm.) | 450s (half-life) | N/R | 14 mutations from mGold; enhanced photostability | [65] |
| mGold2t | Yellow | ~514 | ~527 | 85 (mClover3 norm.) | 445s (half-life) | N/R | 15 mutations from mGold; enhanced photostability | [65] |
| mRuby3 | Red | 558 | 592 | 58 | 349s (half-life) | 4.8 | 21 mutations from mRuby2; brightest monomeric RFP | [66] |
| mClover3 | Green | 506 | 518 | 85 | 80s (half-life) | 6.5 | Improved photostability vs Clover (+60%) | [66] |
| mVenus | Yellow | 514 | 527 | ~84 | 23s (half-life) | N/R | Fast maturation; reduced acid sensitivity vs EYFP | [65] [62] |
| mCherry | Red | 587 | 610 | 16 | 96s (half-life) | <4.5 | Monomeric; widely used | [66] |
| TagRFP-T | Red | 555 | 584 | 33 | 337s (half-life) | 4.6 | High photostability | [66] |
| StayGold | Green | ~506 | ~518 | High | Extremely high | N/R | Metagenomic discovery; benchmark photostability | [65] |
Brightness expressed as product of extinction coefficient (mM⁻¹cm⁻¹) and quantum yield, normalized to mClover3 where applicable for cross-study comparison. *Photostability measured as half-life under widefield illumination at 520nm (YFPs) or similar conditions, when available.
Applications in FRET Biosensor Design
Principles of FRET Biosensors
FRET biosensors typically consist of two FPs with overlapping emission and excitation spectra linked by a sensing domain that changes conformation in response to a biochemical event [63] [64]. When the biosensor is in its inactive state, the FPs are sufficiently close for energy transfer from the donor to acceptor FP upon donor excitation. Upon activation (e.g., by calcium binding, phosphorylation, or protein interaction), conformational separation reduces FRET efficiency, providing a measurable signal change [64] [34]. The critical parameters for FRET pairs include spectral overlap, brightness, photostability, and maturation efficiency.
Advanced FRET Biosensor Designs
Recent innovations in FP engineering have enabled more sophisticated FRET biosensor designs:
- Near-Infrared FRET Biosensors: Utilizing FPs such as miRFP670 and miRFP720, these biosensors enable spectral resolution from existing biosensors in the blue/green/yellow range while reducing cellular autofluorescence [34]. Applications include monitoring small GTPase activity (e.g., Rac1) in real time, providing insights into cancer cell migration and invasion.
- Optimized GFP-RFP Pairs: Pairs such as mClover3-mRuby3 offer advantages over traditional CFP-YFP pairs, including greater spectral separation, less phototoxicity, lower autofluorescence, and improved performance in tissue imaging [66].
- Single FP Biosensors: Utilizing environment-sensitive FPs, these designs incorporate sensing domains within single FPs, changing fluorescence properties in response to analyte binding or environmental changes [63].
Diagram 2: FRET biosensor working principle. Biosensors transition between high-FRET (inactive) and low-FRET (active) states in response to biochemical events, detected as changes in emission ratios. [63] [64] [34]
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Research Reagents for Fluorescent Protein Engineering and Application
| Reagent / Tool | Function / Application | Examples / Specifications | Reference |
|---|---|---|---|
| SPOTlight Platform | High-throughput screening of FP libraries for brightness & photostability | Single-cell analysis; upgraded with mTurquoise2 reference & minimal media | [65] |
| FRET Biosensor Plasmids | Monitoring molecular interactions & enzymatic activities | NIR FRET Rac1 biosensor (miRFP670/720); Camuiα (CaMKIIα activity) | [66] [34] |
| Directed Evolution Kits | Random mutagenesis & library generation | Error-prone PCR kits; site-saturation mutagenesis kits | [65] [66] |
| Spectrophotometry Systems | Characterizing spectral properties & quantum yields | Instruments for measuring extinction coefficients & emission spectra | [65] [66] |
| Advanced Microscopy Platforms | Validating FP performance in physiological contexts | TIRF, super-resolution, confocal systems with environmental control | [65] [64] |
| Subcellular Targeting Vectors | Testing FP localization & artifacts | Organelle-specific tags (nuclear, mitochondrial, plasma membrane) | [66] |
The engineering of fluorescent proteins with enhanced brightness, photostability, and pH resistance has dramatically expanded the capabilities of genetically encoded biosensors. Through rational design, directed evolution, and emerging de novo approaches, researchers have developed FPs that overcome previous limitations in imaging duration, signal intensity, and environmental sensitivity. These advances directly translate to improved FRET biosensors that enable more precise and prolonged observation of dynamic cellular processes, from kinase activity to small GTPase signaling.
Future developments will likely focus on further expanding the color palette, particularly in the near-infrared spectrum for deeper tissue imaging, and creating specialized FPs with orthogonal properties for simultaneous monitoring of multiple signaling pathways. The integration of computational design approaches with high-throughput experimental screening promises to accelerate the development of next-generation FPs tailored for specific biosensing applications, ultimately providing researchers with an evermore sophisticated toolkit for deciphering the complexity of living systems.
In the sophisticated architecture of genetically encoded FRET biosensors, the sensor domain functions as the critical molecular intelligence unit, directly responsible for translating a biochemical event—such as ligand binding, enzymatic activity, or a conformational change—into a quantifiable FRET signal change [60]. This domain's affinity and specificity for its target analyte fundamentally determine the biosensor's performance, sensitivity, and utility in biological research. Engineering this component through rational design and directed evolution has therefore become a central focus in advancing biosensor technology, enabling researchers to probe cellular dynamics with ever-increasing precision [60] [42]. The imperative for such optimization is clear: many biological processes occur at physiologically relevant concentrations that are beyond the detection limits of first-generation biosensors. This technical guide details the core strategies and methodologies for engineering sensor domains, with a specific focus on mutagenesis and affinity tuning, to equip researchers with the tools necessary to develop bespoke biosensors for challenging applications.
Core Mutagenesis Strategies for Affinity and Specificity Optimization
Binding Site Mutagenesis for Enhanced Affinity
A primary and highly effective strategy for tuning biosensor affinity is the targeted mutagenesis of key residues within the sensor domain's binding pocket. This approach aims to alter the intermolecular interactions—such as hydrogen bonding, van der Waals forces, and electrostatic contacts—between the sensor and its target molecule.
A seminal example is the development of the ECATS2 biosensor for extracellular ATP. The first-generation ecATeam3.10 biosensor, which utilizes the ε subunit of bacterial ATP synthase, exhibits micromolar affinity. To enhance sensitivity for physiological ATP release, researchers introduced a double mutation (R103A/R115A) within the ATP-binding site. This strategic alteration resulted in a second-generation biosensor (ECATS2) with a greater than three-fold higher affinity for its ligand. When characterized in solution, the purified double-mutant sensor domain showed a 4-fold increase in ATP affinity, achieving an apparent dissociation constant (K_D) of 0.2 µM, making it capable of detecting more subtle fluctuations in extracellular ATP dynamics [68].
This example underscores a general principle: even a minimal number of mutations, if strategically chosen, can yield significant improvements. The process often begins with structural analysis (e.g., via X-ray crystallography or AlphaFold models) to identify residues that interact with the target or stabilize the binding pocket [68]. Site-saturated mutagenesis of these positions can then be employed to exhaustively explore the mutational space and identify optimal substitutions [69].
Engineering Specificity and Creating Novel Binding Interfaces
Beyond enhancing affinity, mutagenesis is a powerful tool for increasing biosensor specificity or even conferring entirely new binding capabilities. This is particularly valuable when developing sensors for specific post-translational modifications or to distinguish between highly similar isoforms.
The engineering of a monobody into a PEbody illustrates this concept. Researchers started with a monobody scaffold and used site-saturation mutagenesis on its BC and FG loops to create a library of variants. Through iterative rounds of fluorescence-activated cell sorting (FACS) and sequence-function analysis, they evolved a novel binder (PEbody) with high affinity (K_D = 5.7 nM) and specificity for R-Phycoerythrin (R-PE). This engineered binder was subsequently fused with ECFP to create a hybrid FRET biosensor for monitoring protease activity at the live-cell surface, a application for which no natural high-affinity binder existed [70].
Similarly, for epigenetic monitoring, a highly specific FRET biosensor for H3K9 acetylation (H3K9ac) was developed by systematically testing bromodomains from various reader proteins. Optimization through site-saturated mutagenesis yielded a triple-mutant biosensor with a dynamic FRET change of up to 30%, enabling specific detection of this single epigenetic mark without cross-reactivity [69].
Table 1: Representative Examples of Sensor Domain Optimization via Mutagenesis
| Biosensor Target | Sensor Domain | Mutagenesis Strategy | Key Mutation(s) | Affinity/Performance Outcome |
|---|---|---|---|---|
| Extracellular ATP [68] | ATP synthase ε subunit | Structure-informed rational design | R103A, R115A | >3-fold higher affinity; K_D of 0.2 µM |
| R-PE Binder (PEbody) [70] | Monobody scaffold | Directed evolution & site-saturation | FINFK (BC loop), WRWWY (FG loop) | K_D of 5.7 nM; enabled hybrid FRET sensor assembly |
| H3K9 Acetylation [69] | BRD4 bromodomain | Site-saturated mutagenesis | Triple mutant (unspecified) | ~30% dynamic FRET range; high specificity |
| Fyn Kinase [42] | Peptide substrate | FRET-Seq of randomized libraries | Variants in +1, +2, +3 positions | Identified sensitive biosensors with improved dynamic range |
Advanced and High-Throughput Engineering Methodologies
The FRET-Seq Platform for Mammalian Cell Screening
Conventional biosensor engineering is often laborious and low-throughput. The FRET-Seq platform overcomes this by integrating high-throughput FRET-based cell sorting with next-generation sequencing (NGS) directly in mammalian cells [42]. This method is designed to efficiently screen large libraries (e.g., >30,000 variants) for optimal sensor domains.
The workflow involves creating a library of biosensor variants, typically by randomizing the amino acid sequences flanking the target residue in a substrate peptide. This library is expressed in mammalian cells using a self-activating FRET (saFRET) design, where an active kinase domain is fused to the biosensor to ensure phosphorylation and FRET signal generation is intrinsic to the biosensor itself. Cells are then sorted via FACS based on their FRET ratios. The DNA from sorted populations is subjected to NGS, and computational analysis identifies enriched variants based on predefined criteria [42].
This powerful platform was used to optimize biosensors for Fyn and ZAP70 kinases. By randomizing residues around the phosphorylatable tyrosine, researchers identified substrate sequences that led to biosensors with significantly enhanced sensitivity, enabling the monitoring of previously undetectable kinase dynamics in live T cells [42].
Linker Optimization and Structural Rigidification
The performance of a FRET biosensor is not solely dependent on the sensor domain's affinity; the structural coupling of the binding-induced conformational change to the fluorescent proteins is equally critical. The linkers connecting these domains play a vital role in transmitting this change.
Research on the calcium biosensor Twitch-2B provides a textbook example. A high-resolution crystal structure revealed that the linkers between the sensing and fluorescent domains formed specific interactions. However, NMR studies showed significant interdomain dynamics that limited FRET efficiency. To counter this, researchers designed a mutation (N532F) that created a new hydrophobic interaction between the acceptor fluorescent protein (cpVenus) and the sensing domain (TnC). This rigidified the entire biosensor structure, reducing dynamics and increasing the maximal FRET ratio change from 800% to 1100% [71].
Furthermore, the introduction of ER/K linkers (which have a propensity to form α-helices) has been shown to improve the dynamic range of FRET biosensors by providing a more rigid and defined structural connection, thereby more efficiently translating a small conformational change in the sensor domain into a larger change in the distance or orientation between the donor and acceptor fluorophores [9].
Detailed Experimental Protocols
Protocol 1: Structure-Informed Binding Site Mutagenesis
This protocol is ideal for sensor domains with known or predicted structures.
Step 1: Identify Target Residues for Mutagenesis
- Generate a structural model of the sensor domain bound to its target using X-ray crystallography, NMR, or computational tools like AlphaFold [68].
- Analyze the binding interface to identify residues involved in:
- Direct hydrogen bonding or electrostatic interactions with the target.
- Van der Waals contacts within the binding pocket.
- Stabilizing the binding-competent conformation.
Step 2: Design and Generate Mutants
- For rational design, select residues for alanine scanning or charge-reversal mutations to disrupt or enhance specific interactions.
- Use site-directed mutagenesis kits (e.g., NEB Q5 Site-Directed Mutagenesis Kit) to introduce specific point mutations into the biosensor plasmid [68].
- For broader exploration, perform site-saturation mutagenesis on 1-3 key residues to generate a small library of variants.
Step 3: Characterize Mutant Biosensors
- Purify the mutant biosensor proteins and perform in vitro titration with the target molecule.
- Fit the dose-response data to determine the apparent dissociation constant (K_D) and compare it to the wild-type sensor [68].
- For cell-based sensors, transfert the mutants into cultured cells and perform live-cell dose-response assays, calculating the FRET ratio change (ΔR/R) for each ATP concentration [68].
Protocol 2: FRET-Seq for High-Throughput Biosensor Optimization
This protocol is for engineering peptide-based sensor domains (e.g., for kinases) within a complex library.
Step 1: Library Design and Construction
- Design degenerate primers to randomize 4-6 amino acid positions within the substrate peptide sequence (e.g., positions -3 to -1 and +1 to +3 relative to the modified residue) using an NNK codon strategy [42].
- Clone the randomized library into a saFRET biosensor vector, which includes a fused, constitutively active kinase domain.
Step 2: Mammalian Cell Library Generation and FACS
- Generate a high-titer lentivirus from the biosensor library DNA.
- Infect a mammalian cell line (e.g., HEK293) at a low multiplicity of infection (MOI) to ensure most cells express a single variant.
- Use a FACS sorter to collect cell populations with desired FRET ratios (e.g., high FRET for the active kinase library and low FRET for a kinase-dead control library) [42].
Step 3: Next-Generation Sequencing and Data Analysis
- Isolate genomic DNA from the pre-sort (input) and sorted cell populations.
- Amplify the biosensor coding region by PCR and subject the products to NGS.
- Analyze the sequencing data to calculate an enrichment ratio (Ev) for each variant. Desired biosensors are those highly enriched in the active-kinase/high-FRET sort and depleted from control sorts [42].
- Select top-ranked sequences for downstream validation and functional imaging.
The Scientist's Toolkit: Essential Research Reagents
Table 2: Key Reagent Solutions for Sensor Domain Engineering
| Reagent / Tool | Function / Application | Specific Examples / Notes |
|---|---|---|
| Site-Directed Mutagenesis Kits | Introduction of specific point mutations into biosensor plasmids. | NEB Q5 Site-Directed Mutagenesis Kit [68] |
| Gibson Assembly / HiFi Assembly | Seamless cloning of biosensor components and linker swaps. | NEB Gibson Assembly Master Mix [68] |
| ER/K Peptide Linkers | Optimization of structural coupling between domains; enhances dynamic range by reducing flexibility. | Used to improve FRET biosensor performance [9] |
| Mammalian Expression Vectors & Viral Systems | Biosensor delivery and stable expression in mammalian cells for library screening and live-cell imaging. | Adenovirus (Adeno-X) or Lentivirus systems [68] [42] |
| Fluorescence-Activated Cell Sorter (FACS) | High-throughput screening and isolation of cells based on FRET efficiency. | Used in FRET-Seq and directed evolution platforms [70] [42] |
| Biolayer Interferometry (BLI) | Label-free measurement of binding kinetics (KD, kon, k_off) between purified sensor domain and target. | Used to characterize PEbody affinity (K_D = 5.7 nM) [70] |
Visualizing Engineering Workflows and Signaling Pathways
Mutagenesis and Screening Workflow
FRET-Seq Platform Logic
Biosensor Response Signaling Pathway
Förster Resonance Energy Transfer (FRET) biosensors are powerful tools for studying cellular signaling dynamics, but their quantitative potential is often hampered by spectral overlap and background noise. This technical guide details the core principles and practical calibration standards necessary to overcome these challenges. We explore robust measurement methodologies, the advantages of time-resolved detection, and the implementation of novel fluorophore pairs to achieve precise, quantifiable FRET measurements in live cells. By providing standardized protocols and validation frameworks, this whitepaper aims to empower researchers in the design and application of genetically encoded FRET biosensors for advanced biological research and drug discovery.
The fundamental principle of FRET relies on the non-radiative energy transfer from an excited donor fluorophore to a nearby acceptor fluorophore, a phenomenon highly sensitive to molecular proximity (1–10 nm) and orientation [3] [2]. This distance dependence makes FRET an exceptionally powerful tool for monitoring protein-protein interactions, conformational changes, and biomolecular dynamics in living cells with high spatial and temporal resolution [72] [49]. However, the quantitative accuracy of FRET measurements is intrinsically challenged by spectral overlap, where the emission spectrum of the donor fluorophore significantly overlaps with the excitation and emission spectra of the acceptor fluorophore [72]. This overlap leads to two primary artifacts: donor emission crosstalk (where donor fluorescence is detected in the acceptor's emission channel) and acceptor excitation crosstalk (where the acceptor is directly excited by light intended for the donor) [73]. Without rigorous correction, these artifacts can obscure genuine FRET signals, leading to inaccurate estimations of FRET efficiency and, consequently, erroneous biological interpretations.
The push for multiparameter imaging—simultaneously monitoring multiple signaling pathways in a single cell—has further exacerbated these spectral challenges [32] [36]. Traditional FRET pairs, such as CFP/YFP, occupy a large portion of the visible spectrum, limiting the availability of spectral windows for additional biosensors [74]. Furthermore, the performance of FRET biosensors is highly dependent on their dynamic range—the magnitude of signal change between the active and inactive states. Many conventional biosensors suffer from relatively low dynamic ranges, necessitating laborious optimization and screening [74]. This whitepaper addresses these interconnected issues by framing them within the broader context of FRET biosensor design research, providing a comprehensive guide to calibration standards and methodological approaches that ensure robust, quantifiable measurements essential for both basic research and high-throughput drug development.
Core Principles and Measurement Methodologies
Quantitative Foundations of FRET
The efficiency of FRET (E) is quantitatively described by its inverse sixth-power dependence on the distance (r) between the donor and acceptor fluorophores, as defined by the Förster equation: [E = \frac{1}{1 + (r/R0)^6}] Here, (R0) is the Förster radius, the distance at which FRET efficiency is 50% [72] [2]. The value of (R_0) is specific to each fluorophore pair and is determined by the quantum yield of the donor, the relative orientation of the dipoles (κ²), the refractive index of the medium, and most critically, the degree of spectral overlap (J) between the donor's emission spectrum and the acceptor's absorption spectrum [72] [75]. A high degree of spectral overlap is essential for efficient energy transfer but is also the primary source of the crosstalk artifacts that complicate quantification. The orientation factor κ² is particularly tricky; while it is often assumed to be ⅔ in dynamic systems where fluorophores rotate freely, this assumption can be invalid in restricted environments, leading to inaccurate distance calculations [2] [75]. Therefore, understanding and controlling for these parameters is the first step in designing a robust quantitative FRET experiment.
Comparative Analysis of FRET Measurement Techniques
Several methodologies exist for measuring FRET efficiency, each with distinct advantages, limitations, and calibration requirements. The choice of technique depends on the experimental system, the required precision, and the available instrumentation.
Table 1: Key Techniques for Measuring FRET Efficiency
| Method | Principle | Advantages | Limitations | Primary Application |
|---|---|---|---|---|
| Sensitized Emission (Ratiometric) [3] [49] | Measures increase in acceptor emission upon donor excitation. | Technically simple, fast, compatible with live-cell imaging. | Requires rigorous correction for crosstalk and concentration; semi-quantitative without calibration. | High-throughput screening, kinetic studies in live cells. |
| Acceptor Photobleaching [2] | Measures increase in donor fluorescence after bleaching the acceptor. | Conceptually simple; directly calculates E from donor intensity. | Destructive; not suitable for kinetics; requires stable samples. | Validating FRET in fixed cells or at end-points. |
| Fluorescence Lifetime Imaging (FRET-FLIM) [32] [49] | Measures decrease in donor fluorescence lifetime due to FRET. | Insensitive to fluorophore concentration and excitation intensity; highly quantitative. | Requires expensive, specialized equipment; data analysis can be complex. | Most accurate quantification of FRET efficiency in complex cellular environments. |
| Spectral Unmixing (ExEm-spFRET) [73] | Records full excitation-emission spectra and uses linear unmixing to resolve crosstalk. | Inherently corrects for both donor emission and acceptor excitation crosstalk simultaneously. | Data-intensive; requires specialized acquisition systems and analysis algorithms. | Live-cell FRET with superior robustness, especially at low signal-to-noise ratios. |
Among these, the ExEm-spFRET method has demonstrated superior robustness in live-cell measurements. A comparative study showed that while both ExEm-spFRET and the emission-based unmixing method (IIem-spFRET) performed well under high signal-to-noise (S/N) conditions (>9), ExEm-spFRET was significantly more reliable for cells with low S/N ratios (<3), accurately determining FRET efficiency (E) and acceptor-to-donor concentration ratios (RC) where IIem-spFRET failed [73]. This makes it particularly valuable for quantifying weak interactions or for biosensors expressed at low levels.
Advanced Tools and Reagents for Enhanced FRET
The development of novel fluorophores and assay technologies has been instrumental in overcoming the traditional limitations of FRET biosensors.
Research Reagent Solutions
A critical step in experimental design is the selection of appropriate reagents. The following table details key tools and their functions in modern FRET research.
Table 2: Essential Research Reagents for FRET Biosensor Development
| Reagent / Tool | Function / Description | Key Application in FRET |
|---|---|---|
| ChemoG5 FRET Pairs [74] | Engineered fusion of a fluorescent protein (e.g., eGFP) and a HaloTag labeled with a rhodamine fluorophore (e.g., SiR). | Creates chemogenetic FRET pairs with near-quantitative FRET efficiency (≥95%), enabling biosensors with unprecedented dynamic ranges for Ca²⁺, ATP, and NAD⁺. |
| Red-FRET ERK Biosensors (REKAR) [36] | Biosensors using miRFP670nano3 and miRFP720 as a FRET pair, operating in the 670–720 nm range. | Enables multiplexed imaging with traditional CFP/YFP biosensors by eliminating spectral overlap. REKAR67 offers a higher dynamic range, while REKAR76 exhibits lower signal variance. |
| Lanthanide Donors (Eu³⁺, Tb³⁺ Cryptates) [76] | Long-lived fluorescent donors used in Time-Resolved FRET (TR-FRET) like HTRF. | Eliminates short-lived background fluorescence via time-delayed detection, dramatically reducing autofluorescence and scatter interference for high-throughput screening. |
| XL665/d2 Acceptors [76] | Synthetic fluorophores that emit in the red spectrum, paired with lanthanide cryptates. | Serves as the acceptor in HTRF assays. The d2 acceptor is smaller, minimizing steric hindrance in biomolecular binding assays. |
| Circularly Permuted FPs (cpFPs) [75] | Fluorescent proteins with rearranged N- and C-termini, altering the environmental sensitivity of the chromophore. | Used in intensiometric biosensors (e.g., ExRai-AKTAR2) and to optimize the orientation of FPs in FRET biosensors for improved dynamic range. |
The Power of Time-Resolved FRET (HTRF)
Homogeneous Time-Resolved Fluorescence (HTRF) is a variant that combines FRET with time-resolved detection. This technology uses lanthanide cryptates (e.g., Europium Eu³⁺) as donors, which have exceptionally long fluorescence lifetimes (microseconds to milliseconds). By introducing a time delay between excitation and emission detection, short-lived background fluorescence (from the buffer, compounds, or biological samples, which lasts only nanoseconds) is eliminated, resulting in a dramatically improved signal-to-noise ratio [76]. HTRF is a homogeneous, "add-and-read" assay format, making it ideal for automated, high-throughput drug screening applications where robustness and miniaturization are critical.
Expanding the Color Palette for Multiplexing
A major frontier in FRET biosensor design is the creation of spectrally distinct tools for multiplexing. Recent work has produced novel red-FRET biosensors like REKAR67 and REKAR76 for monitoring ERK activity. These biosensors use the fluorescent proteins miRFP670nano3 and miRFP720, which operate in the 670-720 nm range, making them spectrally compatible with existing CFP/YFP FRET biosensors and cpGFP-based biosensors [36]. This allows, for instance, for the simultaneous monitoring of ERK and AKT activity in the same cell—a previously formidable challenge. Furthermore, the ChemoX platform demonstrates how a single design (a FP fused to a HaloTag) can be tuned across the visible spectrum by changing either the FP (creating ChemoB, ChemoC, ChemoY, ChemoR) or the synthetic fluorophore used to label the HaloTag, providing unparalleled flexibility in biosensor design and multiplexing capability [74].
Experimental Protocols for Calibration and Validation
Protocol: Implementing ExEm-spFRET for Live-Cell Measurement
This protocol is adapted from methods demonstrating superior robustness for low signal-to-noise ratio live-cell measurements [73].
- Sample Preparation: Transfert cells with your FRET biosensor construct. Include essential control samples: cells expressing donor-only and acceptor-only constructs.
- Microscope Setup: Use a microscope system capable of acquiring spectral images (e.g., a spectral confocal or widefield system with a tunable filter or spectrometer).
- Spectral Data Acquisition: For each cell (or region of interest), acquire a 3D excitation-emission (ExEm) data cube.
- Excite the sample across a range of excitation wavelengths (e.g., covering both donor and acceptor excitation peaks).
- At each excitation wavelength, collect the full emission spectrum.
- Spectral Unmixing and Calculation: Utilize linear unmixing algorithms to decompose the acquired ExEm spectra into their constituent components (donor emission, acceptor emission, and autofluorescence) based on the reference spectra obtained from the donor-only and acceptor-only controls. This process directly resolves both donor emission crosstalk and acceptor excitation crosstalk, yielding corrected FRET efficiency (E) and acceptor-to-donor concentration ratio (RC) values.
Protocol: Validation via Fluorescence Lifetime Imaging (FRET-FLIM)
FRET-FLIM is considered a gold standard for validation as it is less susceptible to spectral crosstalk and fluorophore concentration [32] [49].
- Instrument Calibration: Calibrate the FLIM system using a sample with a known fluorescence lifetime to ensure accuracy.
- Lifetime Image Acquisition: Acquire fluorescence lifetime images of your cells expressing the FRET biosensor. Focus on the donor channel using pulsed donor excitation and time-gated or time-correlated single-photon counting detection.
- Lifetime Analysis: Fit the fluorescence decay curve for each pixel to determine the donor's lifetime (τ). Compare the lifetime in the presence of the acceptor (τDA) to the lifetime of the donor alone (τD), obtained from the donor-only control sample.
- FRET Efficiency Calculation: Calculate the FRET efficiency (E) using the formula: [E = 1 - \frac{τ{DA}}{τD}] A decrease in the donor's lifetime in the presence of the acceptor is a direct indicator of FRET. Map this efficiency across the cell to visualize spatial activity of your target.
Protocol: HTRF Assay for High-Throughput Screening
This protocol outlines a generic HTRF assay for biochemical interaction screening [76].
- Reaction Setup: In a low-volume 384- or 1536-well microplate, mix the purified binding partners. One partner is labeled with the donor (e.g., Eu³⁺ cryptate), and the other is labeled with the acceptor (e.g., XL665).
- Incubation: Incubate the reaction mixture to allow binding to reach equilibrium. No washing steps are required.
- Time-Resolved Detection: Read the plate on a compatible microplate reader equipped with TR-FRET capabilities.
- Excitation: Use a 337 nm laser or flash lamp.
- Time Delay: Introduce a delay of 50-100 microseconds after excitation.
- Dual-Emission Detection: Simultaneously measure emission at 620 nm (donor signal) and 665 nm (acceptor FRET signal) over a period of 400 microseconds.
- Data Analysis: Calculate the FRET ratio for each well as: (Signal at 665 nm / Signal at 620 nm) * 10⁴. This ratiometric measurement normalizes for well-to-well variability and quenching effects. The resulting ratio is directly proportional to the extent of binding.
Visualizing FRET Biosensor Design and Workflows
Diagram 1: A comprehensive workflow for the design, implementation, and validation of robust FRET biosensors, highlighting critical decision points for overcoming spectral challenges.
Diagram 2: The pathway from the problem of spectral overlap artifacts to the solution through specific calibration and correction methodologies.
The journey toward robust, quantifiable FRET measurements is one of continuous innovation in fluorophore technology, measurement physics, and experimental calibration. The challenges posed by spectral overlap are no longer insurmountable barriers but rather manageable parameters through the disciplined application of the standards and protocols outlined herein. The emergence of chemogenetic pairs with near-quantitative efficiency, robust red-shifted biosensors for multiplexing, and sophisticated analytical methods like ExEm-spFRET and FLIM represent a new era in biosensor design. As the field progresses, the integration of these tools with automated high-throughput platforms and advanced computational analysis, including artificial intelligence [3], will further solidify FRET's role as an indispensable technique for deciphering the intricate dynamics of cellular signaling. The future of FRET lies in its integration into increasingly complex experimental paradigms, allowing researchers to move beyond observing single events to understanding the interconnected network of signaling that governs cell behavior.
Combatting Photobleaching and Environmental Sensitivity for Reliable Long-Term Imaging
Förster Resonance Energy Transfer (FRET) biosensors have become indispensable tools for studying dynamic cellular processes with high spatiotemporal resolution. However, their utility in long-term imaging and quantitative measurements is fundamentally limited by photobleaching and environmental sensitivity. Photobleaching, the irreversible destruction of fluorophores under illumination, leads to signal loss and inaccurate FRET measurements, while environmental factors like pH and solvent polarity can cause unintended fluorescence fluctuations. These challenges are particularly critical in drug development applications where reliable, quantitative data over extended time periods is essential for pharmacological characterization. The finite photostability of fluorescent proteins (FPs) creates a fundamental trade-off between temporal resolution, experiment duration, and signal-to-noise ratio. This technical guide examines the molecular origins of these limitations and presents advanced engineering strategies to overcome them, enabling more robust and reliable biosensing in preclinical research.
Molecular Origins and Impact of Photodegradation
Fundamental Mechanisms of Photobleaching
Photobleaching in FRET biosensors occurs through multiple pathways that permanently diminish fluorescence emission. The primary mechanism involves reactive oxygen species (ROS) generated when fluorophores in excited states interact with molecular oxygen, leading to oxidative damage of the chromophore's chemical structure. This process is exacerbated by high-intensity illumination and prolonged exposure, particularly in live-cell imaging where cellular metabolism produces additional ROS [77]. The vulnerability to photobleaching varies significantly among fluorescent proteins due to differences in their chromophore environments and barrel structures. For instance, conventional CFP-YFP FRET pairs suffer from relatively rapid photodegradation compared to more modern variants, fundamentally limiting their utility in extended kinetic studies [78].
Environmental sensitivity presents another significant challenge, as fluorescence intensity and spectral properties of many FPs are affected by local conditions. The protonation state of the chromophore is highly dependent on pH, with pKa values typically ranging from 6 to 8 for many FPs, causing significant fluorescence fluctuations within physiological pH ranges [79]. Additionally, halide ion sensitivity particularly affects YFP variants, while temperature-dependent maturation and fluorescence intensity further complicate quantitative measurements. These factors collectively introduce substantial noise and systematic errors in FRET efficiency calculations, especially problematic for long-term pharmacological studies where maintaining consistent cellular conditions is challenging [4].
Consequences for Quantitative Biosensing
The practical implications of photobleaching and environmental sensitivity manifest in several critical ways for drug development research. Signal attenuation from photobleaching causes progressive underestimation of FRET efficiency, potentially misinterpreted as biological activity changes. In high-content screening applications, variable photostability creates well-to-well inconsistencies that compromise data quality and statistical power. Furthermore, differential bleaching rates between donor and acceptor fluorophores distort ratiometric measurements, generating artifactual FRET efficiency trends that cannot be corrected through simple normalization [57].
Environmental sensitivity introduces additional confounding factors, as pH variations in different cellular compartments or experimental conditions create spatially and temporally inconsistent biosensor performance. For GPCR activation studies using FRET biosensors, these artifacts can obscure crucial kinetic information about drug-target interactions, particularly problematic when monitoring slow cellular processes or conducting prolonged pharmacological observations [78]. The resulting data variability increases the number of replicates required for statistical significance, raising costs and extending timelines in preclinical drug characterization.
Engineering Strategies for Enhanced Photostability
Advanced Fluorescent Protein Engineering
Protein engineering efforts have yielded fluorescent protein variants with dramatically improved photophysical properties. The development of mTurquoise2 represents a significant advancement, offering a quantum yield of 0.93 and a fluorescence lifetime of 3.8-4.0 ns, substantially higher than conventional CFP variants. Crucially, mTurquoise2 exhibits 3-4 times greater photostability compared to its predecessors, making it an ideal FRET donor for extended imaging sessions [78]. For acceptor fluorophores, circularly permuted Venus (cpVenus) and related variants provide bright emission while maintaining reduced environmental sensitivity, particularly to pH and halide ions. The engineering of tandem dimer acceptors such as tddVenus further enhances signal brightness while minimizing acceptor-donor stoichiometry variations that complicate FRET quantification [57].
Recent breakthroughs in chemogenetic FRET pairs have created entirely new design possibilities. The ChemoX platform engineers a specific interface between a fluorescent protein and a fluorescently labeled HaloTag, achieving near-quantitative FRET efficiencies ≥94% while maintaining exceptional photostability. This system combines the genetic encodability of FPs with the superior photophysical properties of synthetic rhodamine dyes, which typically exhibit higher photostability and lower environmental sensitivity than conventional FPs. The strategic combination of mTurquoise2 donor with spectrally optimized rhodamine acceptors in this platform represents the current state-of-the-art for photostable FRET measurements [35].
Table 1: Photostable Fluorescent Proteins and Their Properties for FRET Biosensors
| Fluorescent Protein | Type/Application | Key Photophysical Properties | Advantages for Long-Term Imaging |
|---|---|---|---|
| mTurquoise2 | Donor FP | Quantum yield: 0.93, Lifetime: ~3.9 ns | 3-4x greater photostability than CFP variants, bright emission |
| cp173Venus | Acceptor FP | Circularly permuted variant | Reduced environmental sensitivity, maintains high quantum yield |
| Tandem dark Venus (tddVenus) | Acceptor FP | Double Venus domains | Enhanced brightness, minimized stoichiometry variations |
| ChemoG5 (with SiR acceptor) | Chemogenetic FRET pair | FRET efficiency: ≥95% | Combines FP genetic encodability with rhodamine photostability |
| Rhodamine-HaloTag conjugates | Synthetic fluorophore | Various emission wavelengths (556-686 nm) | Superior photostability, tunable spectral properties |
Biosensor Architecture and Expression Optimization
Strategic biosensor design extends beyond fluorophore selection to encompass overall architecture and cellular expression control. The implementation of single plasmid systems with viral 2A peptides and IRES sequences ensures optimal donor-acceptor stoichiometry, significantly reducing cell-to-cell variation. This approach maintains a consistent 3:1 expression ratio between upstream and downstream coding sequences, preventing the accumulation of unpaired donors or acceptors that contribute to background noise and photobleaching artifacts [78]. Additionally, optimizing subcellular targeting sequences directs biosensors to appropriate cellular compartments while avoiding regions with potentially damaging environmental conditions, such as acidic organelles.
The engineering of linker sequences between sensor domains and fluorescent proteins also influences photostability by controlling fluorophore separation and orientation. Optimal linkers reduce non-specific fluorophore interactions that might accelerate photodegradation while maintaining the conformational flexibility necessary for biosensor function. Furthermore, selecting maturation-rate matched FP pairs ensures that both donor and acceptor fluorophores become functional simultaneously, preventing periods where unpaired fluorophores are susceptible to degradation before complex formation [79].
Experimental Approaches for Photostability Characterization
Quantitative Photostability Assessment Protocols
Rigorous characterization of FRET biosensor photostability requires standardized experimental protocols that simulate actual imaging conditions. A comprehensive assessment should include continuous illumination tests where biosensor-expressing cells are exposed to constant excitation light at typical imaging intensities while collecting time-lapse fluorescence data. The resulting fluorescence decay curves are fit to exponential functions to extract quantitative photobleaching rate constants, enabling direct comparison between different biosensor variants. This protocol should be performed under physiological conditions (37°C, 5% CO₂) to account for temperature and pH effects on photostability [57].
For environmental sensitivity characterization, a pH titration series from 6.0 to 8.5 determines the pKa of the biosensor response, identifying variants with minimal pH dependence in the physiological range. Similarly, halide sensitivity should be assessed by perfusing cells with solutions containing physiological concentrations of chloride and iodide ions while monitoring fluorescence. These characterization experiments are essential for establishing the operational boundaries within which quantitative FRET measurements remain valid, particularly for mechanopharmacological screening applications where environmental conditions might vary between experiments [4].
Advanced Microscopy Techniques for Reduced Photodamage
Innovative microscopy methodologies significantly extend FRET biosensor utility by minimizing phototoxic effects while maintaining signal quality. Parallelized multi-beam confocal FLIM systems with arrays of 64 beamlets achieve dramatic reductions in peak excitation power (∼1–2 μW per beamlet) while maintaining high temporal resolution (up to 0.5 frames per second). This approach distributes photon collection across multiple beamlets, enabling longer pixel dwell times without increasing photodamage, crucially important for long-term kinetic studies of signaling dynamics [57].
Fluorescence lifetime imaging (FLIM) provides particularly robust FRET quantification by measuring the reduction in donor fluorescence lifetime, a parameter largely independent of fluorophore concentration and excitation intensity. This intensity-independent measurement circumvents many photobleaching artifacts that plague intensity-based FRET measurements. The implementation of rapid lifetime determination methods and phasor analysis approaches further enhances the temporal resolution of FLIM-FRET, making it compatible with dynamic cellular processes while maintaining quantitative accuracy throughout extended imaging sessions [57].
Photostability Assessment Workflow
Implementation Protocols for Robust FRET Imaging
Step-by-Step FLIM-FRET Protocol for Reduced Photobleaching
The following detailed protocol implements multi-beam confocal FLIM-FRET to minimize photobleaching while maintaining quantitative accuracy:
Sample Preparation: Transfert cells with the mTurquoise2-Epac-tddVenus FRET biosensor using a single plasmid system to ensure defined donor-acceptor stoichiometry. Allow 24-48 hours for full biosensor expression and maturation [78].
Microscope Configuration: Configure a multi-beam confocal FLIM system with 64 excitation beamlets and parallel SPAD array detection. Set excitation power to ∼1–2 μW per beamlet at 440 nm excitation wavelength. Adjust detection channels for donor (475±30 nm) and acceptor (535±25 nm) emissions [57].
Acquisition Parameters: Set temporal resolution to 0.5-2 seconds per frame depending on process dynamics. Use a 40× oil immersion objective with high NA (≥1.3) for optimal photon collection. Maintain sample at 37°C and 5% CO₂ throughout acquisition.
Lifetime Data Collection: Acquire time-lapse FLIM data using time-correlated single photon counting (TCSPC). Collect sufficient photons (typically 1000-5000 per pixel) to ensure accurate lifetime determination while minimizing excitation intensity.
Phasor Analysis: Transform fluorescence decay data into phasor plots for visualization and quantitative analysis. Identify positions corresponding to open and closed biosensor conformations based on their characteristic lifetimes [57].
FRET Efficiency Calculation: Calculate FRET efficiency using the formula: EFRET = 1 - (τDA/τD), where τDA is the donor lifetime in the presence of acceptor and τD is the donor lifetime alone (typically 3.92±0.08 ns for mTurquoise2).
This FLIM-based approach provides intensity-independent FRET measurements that remain quantitatively accurate even as partial photobleaching occurs, significantly enhancing reliability for extended duration experiments.
QuanTI-FRET Calibration for Environmental Insensitivity
The Quantitative Three-Image FRET (QuanTI-FRET) method provides robust intensity-based FRET quantification while correcting for environmental sensitivity artifacts:
Three-Image Acquisition: Collect three essential images under defined illumination conditions: IDD (donor channel with donor excitation), IDA (acceptor channel with donor excitation), and IAA (acceptor channel with acceptor excitation) [6].
Correction Factor Determination: Calculate the bleedthrough correction factor (αBT) and direct excitation correction factor (δDE) using a control sample with known donor:acceptor stoichiometry, typically a 1:1 fusion construct.
Efficiency Calculation: Compute the FRET efficiency using the complete equation: E = (IDA - αBTIDD - δDEIAA) / (IDA - αBTIDD - δDEIAA + γIDD) where γ accounts for differences in detection efficiency between channels [6].
Stoichiometry Validation: Simultaneously calculate the stoichiometry parameter S = (IDA - αBTIDD - δDEIAA + γIDD) / (γIDD + IAA/β) to verify data quality, with deviations from expected values indicating potential environmental artifacts or expression problems.
This comprehensive calibration approach yields absolute FRET values that are independent of instrument configuration and robust against minor environmental fluctuations, essential for reproducible data across multiple experimental sessions.
Quantitative FRET Imaging Protocol
Research Reagent Solutions for Enhanced Photostability
Table 2: Essential Reagents for Photostable FRET Biosensor Implementation
| Reagent/Category | Specific Examples | Function/Application | Key Characteristics |
|---|---|---|---|
| Photostable FPs | mTurquoise2, cp173Venus, tddVenus | FRET donor and acceptor elements | High quantum yield, extended photostability, reduced environmental sensitivity |
| Chemogenetic FRET Pairs | ChemoG5 with SiR acceptor | Near-quantitative FRET with synthetic dyes | ≥95% FRET efficiency, superior photostability of rhodamine dyes |
| Expression Systems | Single plasmid with 2A peptides | Controlled biosensor expression | Maintains defined donor:acceptor stoichiometry, reduces cell-cell variation |
| FLIM-FRET Components | SPAD array detectors, TCSPC electronics | Fluorescence lifetime measurement | Intensity-independent FRET quantification, robust to photobleaching |
| QuanTI-FRET Standards | Defined stoichiometry constructs | Calibration of correction factors | Enables absolute FRET measurements independent of instrument |
| Environmental Controls | pH buffers, antioxidant supplements | Minimize environmental artifacts | Stabilize fluorescence during long-term imaging |
The systematic implementation of protein engineering, advanced microscopy techniques, and rigorous calibration methods provides a comprehensive solution to the persistent challenges of photobleaching and environmental sensitivity in FRET biosensors. The strategic combination of photostable fluorescent proteins like mTurquoise2 with chemogenetic approaches that incorporate synthetic fluorophores represents the current state-of-the-art, enabling robust quantitative measurements previously impossible with conventional biosensors. Furthermore, the adoption of intensity-independent detection methods such as FLIM-FRET and comprehensive calibration frameworks like QuanTI-FRET ensures data reliability throughout extended imaging sessions.
Looking forward, several emerging technologies promise further improvements in photostability and environmental robustness. The continued development of ultrastable fluorescent proteins from diverse organisms offers new architectural possibilities, while synthetic biology approaches enabling precise control of biosensor expression levels will minimize cell-to-cell variability. Advanced computational correction algorithms that dynamically model and compensate for photobleaching during acquisition will extend usable experiment durations. Additionally, the integration of machine learning methods for automated quality control and artifact detection will enhance data reliability in high-throughput screening applications. These innovations will collectively advance FRET biosensing capabilities, ultimately providing drug development researchers with more powerful tools for quantifying cellular signaling dynamics with unprecedented accuracy and temporal duration.
Ensuring Reliability: Biosensor Validation, Comparative Analysis, and Benchmarking
Within the field of genetically encoded biosensor research, the development of Förster Resonance Energy Transfer (FRET) biosensors represents a powerful approach for monitoring biochemical events in live cells with high spatiotemporal resolution [3] [4]. However, the transition from a novel biosensor construct to a trusted scientific tool necessitates rigorous validation against established gold standards. This process ensures that the biosensor's readouts accurately reflect biological reality rather than artifacts of the sensing system itself. For FRET biosensors targeting kinase activity, metabolite concentration, or ion dynamics, validation typically involves demonstrating consistency with conventional biochemical assays and previously characterized biosensors [42] [80]. This whitepaper provides a comprehensive technical guide to these critical validation methodologies, offering detailed protocols and analytical frameworks for researchers developing and implementing FRET-based biosensors in drug discovery and basic research.
Fundamental Principles of FRET Biosensors
FRET is a distance-dependent physical process where energy is transferred non-radiatively from an excited donor fluorophore to a nearby acceptor fluorophore [3] [11]. The efficiency of this transfer (E) is highly sensitive to the distance (r) between the donor and acceptor, inversely proportional to the sixth power of the distance:
[E = \frac{1}{1 + (\frac{r}{R_0})^6}]
Here, (R_0) is the Förster radius, characteristic of a specific donor-acceptor pair [4] [9]. Genetically encoded FRET biosensors exploit this sensitivity by coupling the fluorophore pair to a biological sensing element—often a ligand-binding domain or a peptide substrate—such that a conformational change induced by the target analyte alters the distance or orientation between the donor and acceptor, thereby modulating FRET efficiency [4] [42]. This change is typically measured as a ratio of acceptor to donor emission intensity, providing an internal calibration that minimizes artifacts from variations in expression level or excitation intensity [80].
The Critical Need for Validation
Despite their advantages, FRET biosensors can be limited by sensitivity, dynamic range, and off-target effects [42]. A biosensor might fail to report genuine physiological changes due to insufficient affinity for its target, or it might produce signals influenced by unrelated cellular variables such as pH, chloride concentration, or competing enzymatic activities [11]. Validation against gold standards is therefore not merely a supplementary step but a fundamental requirement to establish biosensor fidelity, define its operational limits, and build scientific consensus around its utility [42] [80].
Gold Standard Assays for FRET Biosensor Validation
Correlation with Established Biochemical Methods
The first line of validation involves correlating FRET biosensor signals with measurements from well-characterized, conventional biochemical techniques.
Table 1: Gold Standard Biochemical Assays for Biosensor Validation
| Biochemical Assay | Target Application | Validation Metric | Key Advantages | Notable Limitations |
|---|---|---|---|---|
| Western Blot | Kinase activity, Protein phosphorylation | Correlation of FRET ratio with phospho-specific antibody signal [42] | High specificity, Widely available | Low temporal resolution, End-point measurement |
| Mass Spectrometry | Metabolite concentration (e.g., ATP) | Quantitative comparison of analyte concentration [80] | Highly quantitative, Unbiased detection | Requires cell lysis, Complex sample preparation |
| Enzyme-Linked Immunosorbent Assay (ELISA) | Cytokine secretion, Extracellular ATP | Correlation with secreted or extracellular factors [80] | High sensitivity, Reproducible | Measures extracellular pool, May not reflect intracellular dynamics |
| Luciferase Assays | ATP dynamics | Direct comparison of ATP concentration readings [80] | Extremely high sensitivity for ATP | Biochemical context differs from live-cell FRET |
Experimental Protocol: Validating a Kinase Biosensor via Western Blot
- Cell Stimulation and Imaging: Culture cells expressing the FRET kinase biosensor (e.g., a ZAP70 or Fyn biosensor [42]). Acquire time-lapse FRET ratio images before and after application of a specific agonist (e.g., EGF for Src family kinases) or inhibitor (e.g., PP1).
- Parallel Sample Lysis: At key time points (e.g., baseline, peak FRET response, post-inhibition), rapidly lyse separate sets of stimulated cells to terminate all enzymatic activity.
- Immunoblotting: Perform Western blot analysis on the lysates using antibodies specific for the phosphorylated form of the target kinase's substrate (e.g., anti-phosphotyrosine) and corresponding total protein.
- Data Correlation: Quantify the band intensities from the blots and plot them against the average FRET ratios from the live-cell imaging at the corresponding time points. A strong positive correlation between the FRET ratio and the degree of substrate phosphorylation validates the biosensor's reporting capability [42].
Cross-Validation with Established Biosensors
A powerful validation strategy, especially in live cells, involves co-expression or parallel expression of the novel biosensor with a well-characterized reference biosensor for the same analyte.
Table 2: Cross-Validation with Reference Biosensors
| Reference Biosensor Type | Principle | Validation Approach | Example |
|---|---|---|---|
| Genetically Encoded FRET Biosensor | Ratiometric FRET measurement | Co-imaging and signal correlation in response to identical stimuli [80] | Validating a new extracellular ATP biosensor against ecATeam3.10 [80] |
| Single-color cpGFP Biosensor | Intensity-based change of cpGFP | Parallel recording in cell populations under physiological challenges [80] | Comparing with AT1.03 or AT1.03YEMK for intracellular ATP |
| sniffer cells | Engineered cells reporting on released molecules | Co-culture and monitoring of intercellular signaling events [80] | Validating ATP release from astrocytes |
Experimental Protocol: Cross-Validation of an Extracellular ATP Biosensor
- Sensor Expression: Create two cell populations: one expressing the novel FRET-based extracellular ATP biosensor (e.g., ECATS2 [80]) and another expressing a established sensor like ecATeam3.10 or a luciferase-based reporter.
- Stimulus Application: Apply a known stimulus that evokes ATP release, such as hypoosmotic stress, to both cell populations simultaneously while performing live imaging.
- Dose-Response Analysis: Apply a range of exogenous ATP concentrations (e.g., from nanomolar to millimolar) to both cell populations to compare the affinity ((K_d)), dynamic range, and sensitivity of the two sensors.
- Data Comparison: Plot the response kinetics and dose-response curves of both sensors. A close match in the EC50 values, temporal dynamics, and relative response magnitudes provides strong evidence for the new biosensor's accuracy [80].
Advanced Validation: FRET-Seq for Biosensor Engineering
Recent advances have integrated validation directly into the biosensor development pipeline. The FRET-Seq technology is a prime example, which combines high-throughput FACS sorting with next-generation sequencing to quantitatively identify optimal biosensor variants from large libraries [42].
Diagram 1: FRET-Seq Biosensor Screening Workflow (Title: FRET-Seq Screening and Validation)
Experimental Protocol: The FRET-Seq Validation Pipeline
- Library Construction: Generate a large library (e.g., >30,000 variants) of biosensor sequences by randomizing key residues in the substrate domain. This is done in a "self-activating" FRET (saFRET) construct where an active kinase domain is fused to the biosensor to ensure phosphorylation-driven FRET changes [42].
- Multiplex FACS Sorting: Express the library in mammalian cells and perform FACS sorting to isolate populations with desired FRET ratios under different conditions:
- KAH: High FRET ratio with active kinase.
- KAL: Low FRET ratio with active kinase.
- KDH: High FRET ratio with dead kinase.
- KDL: Low FRET ratio with dead kinase [42].
- Sequencing and Enrichment Analysis: Subject the sorted populations to next-generation sequencing. Calculate an enrichment ratio (Ev) for each variant to identify sequences that are specifically enriched in the KAH and KDL groups, indicating they are sensitive to the active kinase [42].
- Hit Validation: Isolate the top-ranked variants and characterize them in live-cell imaging experiments to confirm improved dynamic range and sensitivity compared to the parental biosensor, thereby validating their superior performance against the library's gold standard [42].
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Reagents for FRET Biosensor Development and Validation
| Reagent / Tool | Category | Function in Validation | Specific Example |
|---|---|---|---|
| Fluorescent Protein Pairs | Biosensor Component | Serve as the donor and acceptor for FRET; choice affects dynamic range and spectral overlap. | CFP-YFP [4], ECFP-YPet [4], mseCFP-mVenus [80] |
| Specific Agonists/Antagonists | Pharmacological Tool | Used to stimulate or inhibit the target pathway to test biosensor specificity and responsiveness. | EGF (for Src kinase [4]), PP1 (Fyn inhibitor [42]), Hypoosmotic Stress (for ATP release [80]) |
| Phospho-specific Antibodies | Biochemical Tool | Enable correlation of FRET signals with direct measurements of phosphorylation via Western Blot. | Anti-phosphotyrosine [42] |
| Adenovirus/Viral Vectors | Delivery System | Ensure efficient and high-level expression of biosensors in primary or hard-to-transfect cells. | Adenovirus for ecATeam expression in astrocytes [80] |
| ER/K Linkers | Protein Engineering Tool | Optimize flexibility and conformational change of the biosensor to enhance dynamic range. | Linkers introduced to overcome limited conformational changes [9] |
| H2B-mApple | Fluorescent Marker | Used as a co-transfection marker to identify biosensor-expressing cells in mixed populations. | Co-transfected with ecATeam3.10 for identification [80] |
Diagram 2: FRET Biosensor Validation Pathways (Title: Biosensor Validation Strategy)
The integration of FRET biosensors into mainstream biological research and drug discovery is contingent upon robust, multi-faceted validation against biochemical gold standards and established technologies. The methodologies outlined—from direct correlation with Western blot analysis to sophisticated high-throughput screening platforms like FRET-Seq—provide a comprehensive framework for establishing biosensor reliability. As the field progresses towards more complex applications, including multiplexed imaging and high-throughput drug screening, the principles of rigorous validation will remain paramount. By adhering to these standards, researchers can confidently deploy FRET biosensors to illuminate dynamic biochemical processes within living systems, thereby accelerating both fundamental scientific discovery and the development of novel therapeutics.
Genetically encoded FRET (Förster Resonance Energy Transfer) biosensors have revolutionized the study of cellular signaling by enabling the real-time monitoring of biological activities with high spatiotemporal resolution. The performance of these biosensors is critically dependent on key parameters such as dynamic range, signal-to-noise ratio (SNR), and signal variance, which collectively determine their utility in basic research and drug development [55] [4]. This whitepaper provides a comprehensive technical analysis of recently developed FRET biosensor variants, comparing their quantitative performance metrics and outlining detailed experimental protocols for their evaluation. Framed within the broader context of biosensor design research, this review synthesizes cutting-edge advancements that are expanding the capabilities of live-cell imaging and high-throughput screening.
Quantitative Performance Comparison of Biosensor Variants
The performance of FRET biosensors is quantified through several key parameters. The dynamic range (also referred to as response or gain) describes the fold-change in FRET ratio between the minimum and maximum activity states of the biosensor [4]. The signal-to-noise ratio (SNR) determines the confidence with which true biological signals can be distinguished from background fluctuations [81]. Variance refers to the consistency of biosensor response across multiple measurements or cellular expressions [36]. Recent engineering efforts have produced biosensors with dramatically improved performance characteristics, as summarized in Table 1.
Table 1: Performance Metrics of Recent FRET Biosensor Variants
| Biosensor Name | Target | FRET Pair | Dynamic Range (Fold-Change) | Signal-to-Noise Ratio | Key Performance Notes | Citation |
|---|---|---|---|---|---|---|
| ChemoG5 | Calcium, ATP, NAD+ | eGFP + HaloTag-SiR | N/A | N/A | Near-quantitative FRET efficiency (95.8±0.1%); Large dynamic range | [74] |
| REKAR67 | ERK activity | miRFP670nano3 + miRFP720 | Higher than REKAR76 | Similar to REKAR76 | Higher dynamic range but greater signal variance than REKAR76 | [36] |
| REKAR76 | ERK activity | miRFP720 + miRFP670nano3 | Lower than REKAR67 | Similar to REKAR67 | Reduced signal variance compared to REKAR67 | [36] |
| hyBRET-ERK | ERK activity | RLuc8-CFP + YFP | Comparable to FRET prototype | N/A | Enables both FRET and BRET readouts; compatible with optogenetics | [82] |
| aSyn-OFP/MFP | α-synuclein oligomerization | mCyRFP1 + mMaroon1 | 2-4x improvement | 2-4x improvement | 3-4x increase in FRET efficiency over GFP/RFP biosensors | [83] |
| PCA Biosensor (Optimized) | Protocatechuic acid | Transcriptional output + GFP | 500-fold | Significantly improved | 30x increase in maximum output; >1500x improved sensitivity | [81] |
The data reveal several key trends in biosensor optimization. The ChemoG5 biosensor platform achieves remarkable performance through a novel engineering approach that creates a reversible interaction between a fluorescent protein and a fluorescently labeled HaloTag, resulting in near-quantitative FRET efficiency of 95.8% [74]. This chemogenetic design enables biosensors for calcium, ATP, and NAD+ with unprecedented dynamic ranges. The spectral properties can be easily tuned by changing either the fluorescent protein or the synthetic fluorophore, allowing simultaneous monitoring of metabolites in different subcellular compartments [74].
The direct comparison of REKAR67 and REKAR76 biosensors demonstrates how fluorophore positioning impacts performance characteristics. While both sensors use the same fluorophores (miRFP670nano3 and miRFP720), their reversed orientation creates measurable differences: REKAR67 displays a higher dynamic range but greater signal variance, whereas REKAR76 shows reduced variance with a more consistent response profile [36]. This tradeoff between dynamic range and variance highlights the importance of biosensor configuration in experimental design.
The aSyn-OFP/MFP biosensor demonstrates how red-shifted FRET pairs can significantly improve performance. This biosensor shows a 3-4 fold increase in FRET efficiency over previous GFP/RFP-based designs, resulting in substantially improved signal-to-noise ratios for detecting α-synuclein oligomerization [83]. Similarly, the hyBRET-ERK platform represents an innovative hybrid approach that maintains the dynamic range of traditional FRET biosensors while enabling additional BRET readouts for applications in optogenetics and whole-animal imaging [82].
Experimental Protocols for Biosensor Characterization
Molecular Engineering of FRET Biosensors
The development of novel FRET biosensors typically begins with the selection of appropriate sensing and reporting domains. For the REKAR biosensors, the fluorophore replacement protocol was used:
- Plasmid Assembly: Replace CFP and YFP fluorophores in the EKAREN4 backbone with miRFP670nano3 and miRFP720 using Gibson Assembly [36].
- Variant Creation: Generate alternative configurations by switching fluorophore positions (REKAR67: miRFP670nano3-miRFP720; REKAR76: miRFP720-miRFP670nano3) [36].
- Control Constructs: Create negative controls (e.g., REKAR67-T/A and REKAR76-T/A) by introducing point mutations (T498A) that abolish kinase activity [36].
For the ChemoG series, the interface engineering protocol was employed:
- Initial Fusion: Fuse eGFP to the N-terminus of HaloTag7 (HT7) to create ChemoG1 [74].
- Stepwise Optimization: Introduce interface mutations (eGFP: A206K, T225R; HT7: E143R, E147R, L271E) to create ChemoG2 through ChemoG5 [74].
- Crystallographic Validation: Confirm fluorophore proximity and interaction interfaces through X-ray crystallography (PDB ID: 8B6T) [74].
Cell Culture and Biosensor Expression
Standardized cell culture protocols are essential for consistent biosensor performance:
- Cell Line Maintenance: Culture MCF-10A cells in appropriate growth medium, passaging before reaching 85% confluency [36].
- Stable Line Generation: Create stable, inducible cell lines using lentiviral transduction and antibiotic selection to minimize cytotoxicity from overexpression [83].
- Transfection Optimization: For transient transfection, use donor:acceptor DNA ratios of 1:8 for single-fusion biosensors (e.g., G-aSyn + aSyn-R) to maximize FRET signal while maintaining sufficient donor expression [83].
- Expression Control: Utilize inducible systems (e.g., tetracycline-inducible promoters) to fine-tune biosensor expression levels and minimize cellular perturbation [83].
Imaging and Data Acquisition
Accurate quantification of biosensor performance requires standardized imaging protocols:
- Microscopy Setup: Use laser-scanning confocal microscopes with 2-photon excitation and time-resolved detection for FLIM-FRET measurements [84].
- Dual-Channel Acquisition: Acquire time series of frames in region of interest with appropriate pixel dwell time to balance sufficient counts for lifetime determination without averaging out sample fluctuations [84].
- Multi-Wavelength Excitation: For red-shifted biosensors (e.g., O/M pairs), utilize both 473-nm and 532-nm laser lines to reduce autofluorescence and provide additional detection channels for excluding interfering compounds [83].
- Environmental Control: Maintain cells at 37°C with 5% CO2 during live-cell imaging to ensure physiological relevance [36].
Data Analysis and Performance Quantification
Robust analytical methods are required to extract performance metrics:
- FRET Ratio Calculation: Calculate the emission intensity ratio (acceptor/donor) for both fluorescence (FRET ratio) and bioluminescence (BRET ratio) [82].
- Fluorescence Lifetime Analysis: Use phasor approach to FLIM as a fit-free method to quantify FRET efficiency in each pixel [84].
- Dynamic Range Calculation: Determine as the ratio of FRET signals between fully activated and basal states [4].
- Signal-to-Noise Assessment: Calculate as the ratio of FRET response magnitude to standard deviation of background measurements [81].
- Variance Quantification: Evaluate consistency of biosensor response across multiple cells and experimental replicates [36].
Biosensor Engineering Workflows and Signaling Pathways
The development and optimization of high-performance FRET biosensors follows systematic engineering workflows that integrate molecular design, cellular implementation, and performance validation. The following diagrams illustrate key processes in biosensor development and function.
FRET Biosensor Engineering Workflow
FRET Biosensor Signaling Mechanism
Essential Research Reagent Solutions
The implementation of FRET biosensor experiments requires specific reagents and tools that enable precise molecular engineering, cellular expression, and quantitative imaging. Table 2 summarizes key research reagent solutions for biosensor development and application.
Table 2: Essential Research Reagents for FRET Biosensor Development
| Reagent Category | Specific Examples | Function and Application | Citation |
|---|---|---|---|
| Fluorescent Proteins | eGFP, YPet, Turquoise2-GL, miRFP670nano3, miRFP720, mCyRFP1, mMaroon1 | FRET pairs for biosensor readout; spectral variants enable multiplexing | [74] [36] [83] |
| Self-Labeling Protein Tags | HaloTag7 (HT7) | Chemogenetic tag for labeling with synthetic fluorophores; enables near-quantitative FRET | [74] |
| Synthetic Fluorophores | Silicon Rhodamine (SiR), Tetramethylrhodamine (TMR), Janelia Fluor dyes (JF525, JF669) | FRET acceptors with superior photophysical properties; spectral tuning | [74] |
| Bioluminescent Proteins | RLuc8 S257G, NanoLuc | BRET donors for low-background imaging and whole-animal applications | [82] |
| Cell Lines | MCF-10A, HEK293, NIH3T3 | Cellular expression systems for biosensor validation and screening | [36] [84] [83] |
| Transfection Reagents | FuGENE HD Transfection Reagent | Efficient delivery of biosensor constructs into mammalian cells | [36] |
| Assembly Kits | Gibson Assembly Master Mix | Molecular engineering of biosensor constructs via seamless DNA assembly | [36] |
| Luciferase Substrates | Coelenterazine-h | BRET substrate for bioluminescence excitation in hybrid biosensors | [82] |
The continuing evolution of FRET biosensor technology is producing increasingly sophisticated tools with enhanced dynamic range, improved signal-to-noise characteristics, and reduced variance. The development of chemogenetic platforms like ChemoG, red-shifted variants such as REKAR67/76, and hybrid systems like hyBRET represents significant advances in biosensor engineering. These tools are expanding our capability to monitor complex signaling networks in live cells and whole organisms with unprecedented precision. As these technologies mature, they promise to accelerate both basic research in cell signaling and applied drug discovery efforts targeting signaling pathway dysregulation. The standardized protocols and performance metrics outlined in this whitepaper provide a framework for the rigorous evaluation and implementation of these powerful molecular tools in research and development settings.
The development of genetically encoded Förster Resonance Energy Transfer (FRET) biosensors represents a cornerstone technology in modern cell biology and drug discovery, enabling the real-time monitoring of biochemical events in living cells with high spatiotemporal resolution. These biosensors typically consist of a sensing domain flanked by donor and acceptor fluorescent proteins, where molecular events such as ligand binding or post-translational modifications induce conformational changes that alter FRET efficiency [3] [8]. However, the mere construction of a FRET biosensor is insufficient to guarantee its scientific utility; rigorous validation of its specificity and functionality is paramount. Without thorough validation, observed FRET changes may result from artifacts rather than genuine biological signals, potentially leading to erroneous conclusions.
Pharmacological and genetic validation approaches serve as complementary strategies to confirm that a biosensor responds specifically to its intended target. Pharmacological validation utilizes chemical inhibitors and activators to modulate the activity of specific pathways, while genetic validation employs techniques such as RNA interference (RNAi), CRISPR/Cas9-mediated gene editing, or overexpression of dominant-negative constructs to manipulate molecular targets [85] [86]. Together, these approaches provide a powerful framework for establishing biosensor specificity, a critical step that must be completed before a biosensor can be reliably deployed to answer biological questions or screen therapeutic compounds. This guide provides researchers with comprehensive methodologies and experimental protocols for implementing these validation strategies effectively.
Fundamental Principles of FRET Biosensor Function
Molecular Mechanisms of FRET Biosensors
FRET biosensors operate on the principle of non-radiative energy transfer between a donor fluorophore and an acceptor fluorophore, a process highly dependent on their proximity (typically within 1-10 nm), relative orientation, and spectral overlap [3] [8]. The efficiency of this energy transfer (FRET efficiency, EFRET) is quantitatively described by the equation:
[E{FRET} = \frac{R0^6}{R_0^6 + R^6}]
Where R represents the distance between donor and acceptor, and R₀ is the Förster radius at which FRET efficiency is 50% [8] [9]. Biosensor design exploits this distance dependence by embedding a sensing domain (e.g., a kinase substrate sequence or ligand-binding domain) between the fluorophores. When the sensing domain undergoes a conformational change upon activation—such as the autophosphorylation of Aurora Kinase A on Thr288 [85] or glucose binding to MglB protein [87]—it alters the distance and/or orientation between the donor and acceptor, resulting in a measurable change in FRET efficiency.
Biosensor Architectures and Their Implications for Validation
The most common biosensor architectures include intramolecular single-chain constructs where donor and acceptor fluorophores are linked by a sensing domain, and intermolecular designs where interactions between separately expressed proteins generate FRET signals. The cpFRET toolkit exemplifies advanced intramolecular designs that employ circularly permuted fluorescent proteins (cpFPs) to optimize the dynamic range by manipulating fluorophore orientation [88]. These designs vary in their topology—"fluorophore-outside" configurations place sensing domains between fluorophores, while "fluorophore-inside" configurations embed fluorophores within the sensing domain [88]. Each architecture presents unique validation considerations, as pharmacological or genetic manipulations must produce predictable and measurable changes in the FRET signal that correspond to the biosensor's structural design and intended biological function.
Pharmacological Validation Strategies
Experimental Design and Protocol
Pharmacological validation utilizes specific chemical compounds to modulate the activity of the target pathway, thereby demonstrating that the biosensor responds predictably to these perturbations. A well-designed pharmacological validation experiment should include both activators and inhibitors of the target pathway to demonstrate that the biosensor signal can be bidirectionally modulated. The following protocol outlines a standardized approach for pharmacological validation:
Step 1: Establish Baseline Biosensor Activity
- Culture cells expressing the biosensor under appropriate conditions (e.g., serum starvation for growth factor pathways).
- Acquire baseline FRET measurements using an appropriate detection method (rationetric imaging, FLIM, or spectral imaging).
- For microscopy, collect at least 10-20 fields of view across multiple biological replicates to establish a robust baseline.
Step 2: Apply Pathway Activators
- Treat cells with a specific activator of the target pathway (e.g., EGF for MAPK/ERK pathway, H₂O₂ for redox sensors).
- Include multiple concentrations to demonstrate dose-dependency (e.g., 0.1, 1, 10, 100 ng/mL for growth factors).
- Perform time-lapse imaging to monitor FRET dynamics immediately following stimulation (e.g., every 30 seconds for 30-60 minutes).
Step 3: Apply Pathway Inhibitors
- Pre-treat cells with specific inhibitors for 30-120 minutes before acquiring baseline measurements.
- Test multiple inhibitor concentrations to demonstrate specificity and establish IC₅₀ values where possible.
- After establishing inhibitor effects, attempt to rescue biosensor activation by adding pathway activators.
Step 4: Control Experiments
- Treat biosensor-expressing cells with compounds targeting unrelated pathways to confirm lack of response.
- Test biosensor response in cell-free systems (purified biosensor protein) to confirm direct mechanism of action.
- Validate compound efficacy using orthogonal assays (e.g., Western blotting for phosphorylation events).
Table 1: Example Pharmacological Agents for Common Pathways
| Target Pathway | Activators | Inhibitors | Expected FRET Response |
|---|---|---|---|
| MAPK/ERK | EGF (10-100 ng/mL), FBS (1-10%) | U0126 (10 µM), PD0325901 (1 µM) | Increase with activation; decrease with inhibition |
| AKT | Insulin (100 nM-1 µM), IGF-1 (50-200 ng/mL) | AKTi VIII (5 µM), MK-2206 (1 µM) | Increase with activation; decrease with inhibition |
| Aurora Kinase A | - | MLN8237 (Alisertib, 100 nM-1 µM) | Decrease with inhibition [85] |
| Calcium Signaling | ATP (100 µM), Carbachol (1 mM), Ionomycin (1-5 µM) | BAPTA-AM (10-50 µM), EGTA (5 mM) | Increase with calcium elevation; decrease with chelation |
Case Study: Validating an Aurora Kinase A FRET Biosensor
A FRET biosensor for Aurora Kinase A (AURKA) was rigorously validated using pharmacological approaches [85]. The biosensor (GFP-AURKA-mCherry) was designed to detect conformational changes induced by autophosphorylation on Thr288. In vitro validation demonstrated that:
- Dephosphorylation using lambda protein phosphatase (λPP) progressively increased the fluorescence lifetime of the donor (reduced FRET) over 35-50 minutes at 30°C.
- Subsequent incubation with ATP triggered re-phosphorylation, decreasing fluorescence lifetime (increased FRET) within 25 minutes.
- Treatment with the specific AURKA inhibitor MLN8237 (Alisertib) prevented the conformational changes associated with activation, confirming that the biosensor response depended on catalytic activity.
This comprehensive approach established a direct link between the biosensor's FRET signal and the phosphorylation state of AURKA, demonstrating its utility for monitoring kinase activity in living cells.
Implementation in High-Throughput Screening
FRET biosensors enable high-throughput pharmacological screening, as demonstrated in triple-negative breast cancer (TNBC) cells [86]. Researchers established a FRET-based imaging approach to monitor ERK and AKT activity simultaneously, then screened a library of >350 kinase inhibitors. This approach revealed differential kinase dependencies between cell lines and identified compounds that suppressed FRET-ERK activity while inhibiting proliferation. Such systematic pharmacological profiling not only validates biosensor specificity but also provides functional insights into signaling network organization and therapeutic vulnerabilities.
Genetic Validation Methodologies
Experimental Design and Protocol
Genetic validation employs molecular biology techniques to directly manipulate the expression or function of the target protein, providing orthogonal evidence for biosensor specificity independent of pharmacological tools. A comprehensive genetic validation strategy should include both loss-of-function and gain-of-function approaches:
Step 1: Loss-of-Function Approaches
- RNA Interference (RNAi): Design 2-3 distinct shRNAs or siRNAs targeting different regions of the gene of interest.
- CRISPR/Cas9: Generate knockout cell lines using sgRNAs targeting essential exons of the gene.
- Dominant-Negative Expression: Express dominant-negative mutants that disrupt normal protein function.
Step 2: Gain-of-Function Approaches
- Wild-Type Protein Overexpression: Express the target protein at high levels to enhance biosensor activation.
- Constitutively Active Mutants: Express mutants that remain in active conformation (e.g., myr-AKT for PI3K/AKT pathway).
- Conditionally Active Systems: Utilize chemically inducible dimerization or optogenetic tools for precise temporal control.
Step 3: Specificity Controls
- Include non-targeting shRNAs/sgRNAs as negative controls.
- Rescue experiments: Re-express RNAi-resistant wild-type cDNA in knockdown cells.
- Test biosensor response to unrelated genetic manipulations.
Table 2: Genetic Manipulation Tools for Biosensor Validation
| Approach | Tool | Key Considerations | Validation Metrics |
|---|---|---|---|
| Knockdown | siRNA, shRNA | Test multiple sequences; optimize transfection efficiency; monitor duration of knockdown | ≥70% reduction in target mRNA/protein; correlated reduction in biosensor response |
| Knockout | CRISPR/Cas9 | Clone isolated single-cell colonies; sequence target locus; monitor compensatory mechanisms | Complete absence of target protein; absence of biosensor activation |
| Dominant-Negative | Kinase-dead mutants, substrate-trapping mutants | Titrate expression level to avoid non-specific effects | Disruption of biosensor activation without affecting expression |
| Overexpression | Wild-type cDNA, Constitutively active mutants | Use inducible systems to control timing of expression | Enhanced or sustained biosensor activation correlating with expression level |
Case Study: Genetic Manipulation in FRET Biosensor Development
The Aurora Kinase A FRET biosensor was functionally validated by demonstrating its ability to replace endogenous kinase function [85]. Researchers showed that the biosensor could rescue phenotypic defects caused by AURKA depletion, establishing its biological functionality beyond mere reporting capability. This stringent validation approach confirmed that:
- The biosensor maintained proper subcellular localization and timing of activation throughout the cell cycle.
- It interacted appropriately with known binding partners such as TPX2 and CEP192.
- It regulated microtubule stability during G1 phase, revealing a previously uncharacterized function for AURKA.
This comprehensive genetic validation provided strong evidence that the biosensor accurately recapitulated the spatial, temporal, and functional characteristics of the endogenous kinase.
Integrated Workflow for Comprehensive Validation
A robust validation strategy integrates both pharmacological and genetic approaches in a sequential manner to build a compelling case for biosensor specificity. The following workflow diagram illustrates the logical relationship between these experimental approaches:
Diagram 1: FRET Biosensor Validation Workflow
Technical Considerations and Troubleshooting
Optimization of Experimental Conditions
Successful validation requires careful optimization of multiple parameters. For pharmacological studies, determine the optimal treatment duration and concentration range through pilot experiments. Consider compound stability, solubility, and potential off-target effects at high concentrations. For genetic manipulations, confirm manipulation efficiency through qRT-PCR or Western blotting and ensure adequate expression of biosensors and manipulated genes. Cell-type specific differences may necessitate optimization of protocols for different model systems.
Addressing Common Validation Challenges
Several technical challenges commonly arise during biosensor validation:
- Incomplete pathway inhibition: Use multiple inhibitors with different mechanisms of action to confirm results.
- Compensatory mechanisms: Combine pharmacological and genetic approaches to circumvent cellular adaptation.
- Non-specific biosensor effects: Include control biosensors with inactivating mutations in the sensing domain.
- Cellular toxicity: Monitor cell viability throughout experiments and include viability controls.
- Signal-to-noise issues: Optimize biosensor expression levels to avoid artifacts from overexpression.
Data Interpretation and Quantification
Robust quantification of FRET changes is essential for reliable validation. Fluorescence Lifetime Imaging Microscopy (FLIM) provides the most quantitative FRET measurements, as demonstrated in the AURKA biosensor validation where a ~150 ps lifetime change corresponded to approximately 16% FRET efficiency [85]. Rationetric imaging requires careful correction for bleed-through and cross-talk, while intensity-based measurements must account for photobleaching and variations in expression level. Normalize FRET responses to positive and negative controls included in each experiment, and report both effect size and statistical significance.
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Research Reagents for FRET Biosensor Validation
| Reagent Category | Specific Examples | Function in Validation | Key Considerations |
|---|---|---|---|
| FRET Biosensor Kits | cpFRET Biosensor Toolkit [88] | Provides optimized biosensor scaffolds with varied linkers and circularly permuted FPs | Enables rapid screening of biosensor configurations for optimal dynamic range |
| Protein Manipulation Tools | Lambda protein phosphatase (λPP) [85] | Dephosphorylates biosensor to establish baseline FRET state | Requires specific temperature conditions (30°C optimal) for full activity |
| Validated Chemical Inhibitors | MLN8237 (AURKA inhibitor) [85], U0126 (MEK inhibitor) [86] | Specifically inhibit target kinases to demonstrate biosensor specificity | Dose-response essential to establish correlation with FRET signal changes |
| Fluorescent Protein Variants | mTurquoise2/Venus FRET pair [87], GFP/mCherry pair [85] | Provide donor-acceptor pairs with optimized spectral properties | mTurquoise2/Venus offers improved pH and ion stability compared to CFP/YFP |
| Immobilization Systems | HaloTag [35] [87] | Enables purification and stabilization of biosensors for in vitro validation | Critical for applications requiring enhanced sensor stability during long measurements |
| Genetic Manipulation Tools | CRISPR/Cas9 systems, RNAi constructs | Enable knockout/knockdown of target proteins to test biosensor dependency | Essential for establishing specificity through loss-of-function approaches |
Pharmacological and genetic validation represents an indispensable component of FRET biosensor development, providing the experimental evidence necessary to establish specificity and functionality. Through the strategic application of chemical inhibitors, activators, and genetic manipulations, researchers can build a compelling case that their biosensor accurately reports on the intended biological process. The protocols and methodologies outlined in this guide provide a framework for comprehensive validation that addresses the rigorous standards required for basic research and drug discovery applications. As FRET biosensor technology continues to evolve, incorporating advanced designs such as chemogenetic FRET pairs with near-quantitative efficiency [35] and multiplexed imaging capabilities, the importance of robust validation strategies will only increase, ensuring that these powerful tools yield biologically meaningful insights into cellular signaling dynamics.
Genetically encoded FRET biosensors have revolutionized the study of cellular signaling dynamics by enabling real-time monitoring of biochemical events in living systems with high spatiotemporal resolution. The efficacy of these biosensors in complex biological environments is governed by a well-defined set of performance metrics that researchers must carefully optimize and benchmark. This technical guide provides a comprehensive framework for evaluating FRET biosensor performance, detailing critical quantitative parameters, experimental methodologies for their assessment, and advanced reagent solutions that enhance sensor capability. By establishing standardized benchmarking protocols, we empower researchers to develop and select optimal biosensors for specific applications in basic research and drug development, ultimately accelerating our understanding of cellular physiology and signaling pathway dysfunction.
Förster Resonance Energy Transfer (FRET) is a distance-dependent quantum mechanical phenomenon involving non-radiative energy transfer from an excited donor fluorophore to a suitable acceptor fluorophore through dipole-dipole coupling [3] [79]. This process occurs efficiently only when fluorophores are in close proximity (typically 1-10 nm), making FRET an exceptionally powerful molecular ruler for probing protein interactions and conformational changes in live cells [79] [89]. Genetically encoded FRET biosensors incorporate this physical principle into single-polypeptide constructs that transduce biochemical events into measurable fluorescence changes, enabling researchers to visualize signaling molecule activities with subcellular resolution [90] [4].
The fundamental architecture of a FRET biosensor typically consists of five key domains: a FRET acceptor fluorescent protein, a specific binding or sensing domain, a structural linker, a FRET donor fluorescent protein (or self-labeling protein domain), and the target protein of interest [90]. In the context of genetically encoded biosensors, the sensing mechanism relies on a conformational change induced by binding of a target molecule or post-translational modification, which alters the distance and/or orientation between the donor and acceptor fluorophores, thereby modifying FRET efficiency [90] [4]. This change in FRET efficiency produces a corresponding alteration in the emission ratios of the donor and acceptor, providing a ratiometric readout that is largely independent of biosensor concentration and photobleaching [4].
Critical Performance Metrics for FRET Biosensors
Dynamic Range and Signal-to-Noise Ratio
The dynamic range represents the maximum observable signal difference between the active and inactive states of a biosensor and is arguably the most critical parameter determining biosensor sensitivity [4]. Computationally, dynamic range is defined as the range of FRET ratio values across all possible states of the biosensor, typically expressed as a percentage change: [(Rmax - Rmin)/Rmin] × 100%, where R represents the acceptor-to-donor emission ratio [4]. A high dynamic range is essential for compensating the generally low signal-to-noise ratio inherent in intensity-based FRET measurements and enables robust detection of subtle biological changes amidst cellular autofluorescence and instrumentation noise [90]. Recent biosensor engineering efforts have achieved remarkable improvements in dynamic range, with certain lanthanide-based FRET (LRET) biosensors incorporating extended ER/K helical linkers demonstrating up to 1100% dynamic range increases [90].
FRET Efficiency and Förster Radius
FRET efficiency (E) quantifies the proportion of donor excitation events that result in energy transfer to the acceptor rather than donor fluorescence emission or non-radiative decay [79]. This parameter follows an inverse sixth-power relationship with the distance between fluorophores (r): E = 1/[1 + (r/R₀)⁶], where R₀ represents the Förster radius [79]. The Förster radius is the characteristic distance at which FRET efficiency is 50% and is determined by the spectral overlap between donor emission and acceptor absorption, the quantum yield of the donor, the extinction coefficient of the acceptor, and the relative orientation of the fluorophore dipoles [79]. Recent advances in chemogenetic FRET pairs have achieved near-quantitative FRET efficiencies exceeding 94-95% through engineered interfaces between fluorescent proteins and rhodamine-labeled HaloTag [35].
Sensitivity and Specificity
Sensitivity in FRET biosensors operates on two distinct levels: molecular sensitivity refers to the minimum detectable concentration of the target analyte, while conformational sensitivity relates to the ability to detect small structural changes in the sensing domain [4]. In practical terms, sensitivity is often defined as the concentration of stimulant required to increase the FRET ratio to 50% of the dynamic range [4]. Specificity encompasses the biosensor's ability to respond exclusively to its intended target without cross-reactivity with similar biological molecules or pathways. This parameter is primarily determined by the molecular recognition properties of the sensing domain, though fluorophore properties and biosensor design can influence off-target responses to environmental variables such as pH and halide ion concentration [89].
Temporal Resolution and Kinetic Parameters
Temporal resolution defines the minimum time interval over which a meaningful biosensor response can be detected and is governed by the kinetics of the sensing domain conformation change, fluorophore maturation time, and signal-to-noise characteristics [88]. For monitoring rapid signaling events such as calcium transients or kinase activation, biosensors must exhibit response timescales compatible with the biological process of interest. The kinetic parameters of association (kon) and dissociation (koff) rates determine how quickly a biosensor can track dynamic changes in target activity, with faster kinetics enabling detection of more rapid biological fluctuations [88]. Mismatched maturation rates between donor and acceptor fluorescent proteins can compromise FRET measurements, as incomplete chromophore formation in either partner reduces effective FRET efficiency [79].
Table 1: Key Performance Metrics for FRET Biosensor Evaluation
| Metric | Definition | Optimal Range | Measurement Method |
|---|---|---|---|
| Dynamic Range | Maximum signal difference between active and inactive states | >50% (conventional FP-FRET); >500% (LRET/advanced designs) | Fluorometry; microscopy ratio imaging |
| FRET Efficiency | Proportion of donor excitation energy transferred to acceptor | >30% for robust detection; >90% for near-quantitative pairs | Acceptor photobleaching; fluorescence lifetime imaging |
| Sensitivity | Analyte concentration producing half-maximal response | Dependent on biological context; should match physiological relevant concentrations | Dose-response curves with known analyte concentrations |
| Signal-to-Noise Ratio | Ratio of true signal to background noise | >5:1 for reliable detection | Comparison of signal variance in presence vs. absence of stimulus |
| Temporal Resolution | Minimum time for accurate response detection | Compatible with biological process (milliseconds to minutes) | Time-lapse imaging after rapid stimulation |
| Specificity | Selectivity for target vs. similar molecules | Minimal cross-reactivity with related analytes | Challenge with structurally similar compounds; pathway inhibitors |
Quantitative Benchmarking Data and Comparisons
Rigorous benchmarking of FRET biosensors requires quantitative comparison across multiple design parameters and biosensor architectures. The data presented in Table 2 enables researchers to contextualize biosensor performance against state-of-the-art implementations and select appropriate designs for specific applications.
Table 2: Performance Comparison of Advanced FRET Biosensor Architectures
| Biosensor Architecture | FRET Pair | Dynamic Range | FRET Efficiency | Key Applications | Notable Features |
|---|---|---|---|---|---|
| ER/K Linker-Based LRET Biosensor | Tb(III)/EGFP | Up to 1100% with 30nm linker | N/R | Rac1 inhibition assays in cell lysates | Enhanced sensitivity with helical linkers; time-gated detection |
| Chemogenetic FRET Pairs (ChemoG5) | eGFP-SiR HaloTag | N/R | 95.8% ± 0.1% | Calcium, ATP, NAD+ sensing | Near-quantitative efficiency; spectral tunability |
| cpFRET Biosensor Toolkit | cp-mTFP1/cp-Venus | Variable by permutation (40-150% typical) | N/R | ERK signaling; customizable for various targets | Systematic optimization via circular permutation |
| Single-Chain Rac1 Biosensor | mCerulean/YPet | Up to 125% with ER/K linkers | N/R | Live-cell Rac1 activation imaging | Plasma membrane localization via CAAX box |
| Protease Cleavage Sensor | Cerulean/Venus | Very high (complete abolition upon cleavage) | N/R | Caspase activity; apoptosis studies | Binary readout; validation of FRET pairs |
The performance variation across different biosensor architectures highlights the importance of matching design strategy to experimental needs. LRET biosensors with lanthanide donors offer exceptional dynamic range beneficial for screening applications, while chemogenetic pairs provide unparalleled FRET efficiency for demanding detection scenarios [90] [35]. The cpFRET toolkit exemplifies a systematic approach to biosensor optimization through comprehensive sampling of fluorophore orientations and distances [88].
Performance Factor Relationships
Experimental Protocols for Biosensor Validation
Determining Dynamic Range and Calibration
Accurate determination of dynamic range requires experimental conditions that definitively establish the fully active and fully inactive biosensor states. For a Rac1 biosensor, this involves transfection into an appropriate cell line followed by treatment with known activators (e.g., EGF or PDGF) and inhibitors (e.g., NSC23766) to establish the maximum response range [90]. The protocol should include:
- Cell Preparation and Transfection: Plate cells in appropriate imaging chambers and transfect with biosensor plasmid using standard methods (lipofection, electroporation, or viral transduction). Allow 24-48 hours for biosensor expression and maturation.
- Baseline Acquisition: Acquire donor and acceptor images using appropriate filter sets under basal conditions. Calculate baseline FRET ratio (acceptor emission/donor emission).
- Activation/Inhibition: Apply saturating concentrations of activator to achieve fully active state, followed by inhibitor to establish fully inactive state. For intracellular targets, consider co-expression of constitutive active or dominant negative mutants.
- Ratio Calculation and Dynamic Range Determination: Calculate FRET ratios for each state: Dynamic Range = [(Ractive - Rinactive)/Rinactive] × 100%.
For in vitro validation, purify biosensor protein and measure fluorescence spectra in controlled buffer conditions with defined concentrations of target analyte to generate calibration curves [90].
FRET Efficiency Measurements via Acceptor Photobleaching
Acceptor photobleaching provides a direct method for quantifying FRET efficiency by comparing donor fluorescence before and after irreversible bleaching of the acceptor fluorophore [79] [89]. The detailed protocol includes:
- Pre-bleach Imaging: Acquire donor and acceptor channel images using appropriate exposure conditions to avoid saturation.
- Region of Interest Selection: Identify specific cellular regions expressing the biosensor for selective bleaching.
- Acceptor Photobleaching: Expose selected regions to high-intensity illumination at the acceptor excitation wavelength until acceptor fluorescence is reduced by >80%.
- Post-bleach Imaging: Re-acquire donor and acceptor channels using identical settings to pre-bleach imaging.
- Efficiency Calculation: Calculate FRET efficiency as: E = 1 - (FDpre/FDpost), where FD represents donor fluorescence intensity.
This method directly measures the dequenching of the donor fluorophore resulting from elimination of energy transfer, providing a robust quantification of FRET efficiency independent of biosensor concentration [79].
Specificity and Cross-Reactivity Testing
Comprehensive specificity validation ensures that biosensor responses genuinely reflect target activity rather than off-target effects or environmental influences:
- Pharmacological Profiling: Expose biosensor-expressing cells to a panel of pathway-specific agonists and antagonists to confirm expected response patterns.
- Genetic Validation: Employ RNA interference or CRISPR-based approaches to knock down target molecules and confirm abolished biosensor response.
- Environmental Controls: Test biosensor response to potential confounding factors including pH variations, oxidative stress, and osmotic changes.
- Related Analyte Challenge: For metabolite biosensors, test response to structurally similar compounds at physiological concentrations.
Document the half-maximal effective concentration (EC50) for target activation and ensure minimal response to non-target stimuli at equivalent concentrations [4].
Biosensor Validation Workflow
Advanced Research Reagent Solutions
The expanding toolkit of specialized reagents has dramatically enhanced FRET biosensor capabilities, enabling researchers to overcome traditional limitations in dynamic range, spectral compatibility, and detection sensitivity.
Table 3: Essential Research Reagents for FRET Biosensor Development and Application
| Reagent Category | Specific Examples | Function and Utility | Application Notes |
|---|---|---|---|
| Fluorescent Protein Pairs | mCerulean/YPet; cp-mTFP1/cp-Venus; Cerulean/Venus | FRET donor-acceptor combinations with optimized spectral overlap | cpFRET toolkit enables systematic optimization via circular permutation [88] |
| Chemogenetic FRET Systems | ChemoG5 (eGFP-HaloTag interface); JF525-JF669 rhodamines | Engineered interfaces enabling near-quantitative FRET efficiency | Spectral tuning via HaloTag fluorophore labeling; >94% efficiency [35] |
| Lanthanide-Based Donors | Tb(III) complexes with eDHFR tagging domain | Long-lifetime donors for time-gated detection eliminating autofluorescence | Enables LRET with >1000% dynamic range in plate assays [90] |
| Structural Linkers | ER/K α-helical linkers (10nm, 20nm, 30nm lengths) | Control distance and orientation between FRET pairs | Enhanced dynamic range with increased length; 30nm linker showed 1100% range [90] |
| Biosensor Toolkits | cpFRET biosensor kit (Addgene #1000000021) | Pre-designed biosensor backbone for rapid customization | 50 plasmid variants for systematic optimization [88] |
| Expression Systems | pTriEx vectors (bacterial, insect, vertebrate) | Multi-host expression system for in vitro and cellular studies | Enables biosensor validation across different expression platforms [88] |
The strategic selection and combination of these advanced reagents empowers researchers to tailor FRET biosensors for specific experimental needs. The cpFRET toolkit exemplifies a systematic approach to biosensor optimization, providing 50 distinct configurations that vary fluorophore distance through different linker lengths and dipole orientation through circular permutations of both donor and acceptor fluorescent proteins [88]. Meanwhile, chemogenetic systems represent a paradigm shift in FRET pair design, achieving near-quantitative energy transfer through engineered interfaces between fluorescent proteins and synthetic fluorophore-labeled HaloTag [35].
The systematic benchmarking of FRET biosensor performance using standardized metrics and validation protocols is essential for advancing their application in complex biological systems. As biosensor designs continue to evolve toward higher dynamic ranges, improved specificity, and enhanced temporal resolution, their utility in deciphering complex signaling networks and accelerating drug discovery will expand correspondingly. The integration of novel engineering approaches—including chemogenetic FRET pairs, lanthanide-based detection, and systematic optimization toolkits—promises to overcome current limitations and enable previously impossible biological measurements. By adhering to rigorous performance assessment standards and leveraging the expanding reagent toolkit, researchers can develop and implement FRET biosensors with confidence in their quantitative capabilities, ultimately driving new discoveries in cellular signaling and therapeutic development.
Fluorescence Resonance Energy Transfer (FRET)-based biosensors represent a powerful technology for visualizing cellular dynamics and bioenergetics in real time, at subcellular resolutions, and in time scales of seconds [91] [92]. These biosensors operate on the principle of Förster resonance energy transfer, a process involving radiation-less transfer of energy from a "donor" fluorophore to an "acceptor" fluorophore, which is highly sensitive to changes in distance and orientation between the two molecules [91]. The integration of in silico design approaches, particularly molecular dynamics (MD) simulations, has revolutionized biosensor development by enabling researchers to predict and optimize biosensor behavior before embarking on costly and time-consuming experimental procedures [93]. This computational paradigm is especially valuable in the context of genetically encoded biosensors, where rational design can be challenging due to the complex relationship between protein structure, function, and fluorescence output.
The power of FRET-based biosensors lies in their ability to monitor diverse molecular events, including protein-protein interactions, protein-DNA interactions, and protein conformational changes in living cells [91]. Recent applications have extended from basic biological research to biomedical disciplines, including clinical diagnostics, as demonstrated by the development of FRET biosensors for detecting SARS-CoV-2 in biological fluids [93]. Despite their diverse applications, FRET-based sensors still face significant challenges, including the need for higher fluorescence resolution, improved specificity, and more affordable reagents for medical diagnostics [91]. Computational approaches directly address these challenges by enabling precise optimization of biosensor components at the atomic level.
Table 1: Key Characteristics of FRET-Based Biosensors
| Characteristic | Description | Biological Application |
|---|---|---|
| Detection Mechanism | Radiation-less energy transfer between donor and acceptor fluorophores | Monitoring protein-protein interactions, conformational changes |
| Temporal Resolution | Seconds to milliseconds | Real-time monitoring of cellular signaling events |
| Spatial Resolution | Subcellular | Compartment-specific analysis of molecular events |
| Readout Method | Ratiometric imaging, fluorescence lifetime imaging | Quantitative analysis of molecular dynamics |
Computational Framework for Biosensor Design
Molecular Dynamics Simulations in Biosensor Development
Molecular dynamics (MD) simulations provide an atomic-level view of biosensor behavior by calculating the time-dependent evolution of a molecular system according to classical equations of motion. This approach is particularly valuable for understanding the dynamic flexibility of linker regions and their impact on FRET efficiency [93]. In recent implementations, such as the SARS-CoV-2 spike protein detection biosensor, MD simulations were employed to analyze fluorophore behavior and stable FRET efficiency, which are essential for reliable detection [93]. The simulations specifically assessed Green Fluorescent Protein (GFP) stability, confirming minimal unfolding tendencies that could compromise sensor function [93].
The process typically begins with system setup, where the biosensor is solvated in an appropriate water model and ions are added to simulate physiological conditions. This is followed by energy minimization to remove steric clashes, equilibration phases to stabilize temperature and pressure, and finally production runs where trajectory data is collected for analysis. For the SARS-CoV-2 biosensor, MD simulations were crucial for validating the sensor's functioning mechanism by demonstrating that the designed linkers maintained optimal flexibility and stability throughout the simulation period [93]. This computational validation provided critical insights before experimental testing, accelerating the development timeline.
Linker Design and Optimization
Linker regions play a critical role in FRET biosensor performance, as they determine the relative orientation and separation distance between donor and acceptor fluorophores. Computational linker design involves evaluating multiple candidate sequences to identify those with optimal biophysical properties. In the SARS-CoV-2 biosensor development, researchers designed and evaluated specific linkers including AAASSGGGASGAGG, selected for its balance between flexibility and stability, and LEAPAPA, chosen for its minimal structural impact on the interaction elements [93].
MD simulations enable quantitative assessment of linker performance through metrics such as root mean square deviation (RMSD), which measures structural stability over time, and root mean square fluctuation (RMSF), which identifies regions of flexibility. These analyses help predict how linkers will behave in physiological conditions and whether they will maintain the appropriate distance and orientation between fluorophores for optimal FRET efficiency. The selection of AAASSGGGASGAGG for the SARS-CoV-2 biosensor was based on its performance in MD simulations, where it demonstrated a favorable balance between the rigidity needed to maintain biosensor architecture and the flexibility required for analyte-induced conformational changes [93].
Integrated Workflow for Computational Design and Validation
The development of genetically encoded FRET biosensors follows a structured workflow that integrates computational and experimental approaches. This begins with biosensor conceptualization, where the sensing mechanism and structural components are defined, followed by in silico design of individual components, particularly the critical linker regions. The computational validation phase employs MD simulations to assess biosensor stability, flexibility, and predicted FRET efficiency before moving to experimental testing.
Diagram 1: Computational Design Workflow
This iterative process allows for continuous refinement of biosensor designs based on computational insights. For instance, if MD simulations reveal excessive flexibility in a linker region that would compromise FRET efficiency, the design can be modified and re-simulated before experimental validation. This approach significantly reduces development time and resources by identifying promising candidates and eliminating poor performers early in the development pipeline. The workflow emphasizes the complementary relationship between computational predictions and experimental validation, with each informing and refining the other.
Key Experimental Protocols and Methodologies
Molecular Dynamics Simulation Protocol
System Preparation: Begin by constructing the initial biosensor structure using homology modeling or de novo design. Place the biosensor in a simulation box with appropriate dimensions, ensuring sufficient clearance (typically ≥10 Å) from box edges. Solvate the system using water models such as TIP3P or SPC/E, and add ions to achieve physiological concentration (150 mM NaCl) and neutralize system charge [93] [94].
Energy Minimization and Equilibration: Perform energy minimization using steepest descent or conjugate gradient algorithms (500-1000 steps) to remove steric clashes. Subsequently, conduct equilibration in two phases: (1) NVT ensemble (constant Number of particles, Volume, and Temperature) for 100-500 ps to stabilize temperature at 300 K using thermostats such as Berendsen or Nosé-Hoover; (2) NPT ensemble (constant Number of particles, Pressure, and Temperature) for 100-500 ps to stabilize pressure at 1 bar using barostats such as Parrinello-Rahman [94].
Production Run and Analysis: Execute production MD simulation for a time scale sufficient to capture relevant biosensor dynamics (typically 50-200 ns). Use integration time steps of 2 fs, with bonds involving hydrogen atoms constrained using algorithms such as LINCS. Analyze trajectories using RMSD, RMSF, and interatomic distances to assess biosensor stability and conformational dynamics. Calculate FRET efficiency based on donor-acceptor distances using Förster theory [93].
Table 2: Key Parameters for MD Simulations of FRET Biosensors
| Parameter | Typical Settings | Rationale |
|---|---|---|
| Simulation Duration | 50-200 nanoseconds | Sufficient to capture linker flexibility and biosensor dynamics |
| Temperature Control | 300 K, Nosé-Hoover thermostat | Maintain physiological temperature |
| Pressure Control | 1 bar, Parrinello-Rahman barostat | Maintain physiological pressure |
| Time Step | 2 femtoseconds | Balance between computational efficiency and numerical stability |
| Water Model | TIP3P, SPC/E | Accurate representation of solvation effects |
FRET Efficiency Calculations
Theoretical Framework: FRET efficiency (E) can be calculated from MD trajectories using the formula based on Förster theory: E = 1 / [1 + (R/R₀)⁶], where R is the distance between donor and acceptor fluorophores, and R₀ is the Förster radius at which FRET efficiency is 50% [93]. The Förster radius depends on the spectral properties of the fluorophore pair and their relative orientation, represented by the orientation factor κ² [93].
Quenching Efficiency: For biosensors employing quenching mechanisms, calculate quenching efficiency based on Förster energy transfer theory to validate sensor sensitivity [93]. This approach was successfully implemented in the SARS-CoV-2 biosensor, where quenching efficiency calculations validated the sensor's sensitivity before experimental testing [93].
Research Reagent Solutions
Table 3: Essential Research Reagents for FRET Biosensor Development
| Reagent/Category | Specific Examples | Function in Biosensor Development |
|---|---|---|
| Fluorophores | Green Fluorescent Protein (GFP) variants, CFP, YFP | Donor and acceptor pairs for FRET detection [92] [93] |
| Computational Software | Schrödinger Suites, Molecular Dynamics packages | Energy minimization, docking, and simulation [94] |
| Expression Systems | E. coli expression strains | Recombinant biosensor production [93] |
| Linker Sequences | AAASSGGGASGAGG, LEAPAPA | Connect sensor domains with optimal flexibility [93] |
Validation and Case Study: SARS-CoV-2 Detection Biosensor
A recent successful implementation of this integrated approach resulted in the development of an intramolecular FRET biosensor for detecting SARS-CoV-2 in biological fluids [93]. This biosensor incorporated two reporter elements at the N-terminus and C-terminus, with interaction elements mediating their separation, supporting two fluorescence measurement methods: direct measurement and the molecular beacon approach [93].
The computational design phase involved in silico design of linkers using MD simulations to ensure optimal flexibility and stability. The selected AAASSGGGASGAGG linker provided an optimal balance between flexibility and stability, while the LEAPAPA linker demonstrated minimal structural impact on the interaction elements [93]. Fluorophore behavior analysis through MD simulations showed stable FRET efficiency, which is essential for reliable detection [93]. Quenching efficiency calculations based on Förster energy transfer theory further validated the sensor's sensitivity [93].
The biosensor was successfully produced in E. coli, and functional validation demonstrated its ability to detect the Spike protein, with fluorescence recovery proportional to protein concentration [93]. This case study demonstrates how the integrated computational-experimental approach enables rapid development of sensitive and specific biosensors for clinical applications, with the modular computer-aided design facilitating sensitivity optimization [93].
Diagram 2: Biosensor Validation Workflow
Advanced Applications and Future Directions
The integration of MD simulations with FRET biosensor design continues to evolve, with several promising applications and future directions emerging. One significant advancement is the ability to model complex cellular environments, including crowded intracellular conditions that affect biosensor performance. Future developments may incorporate machine learning algorithms to predict optimal biosensor configurations based on training data from previous MD simulations and experimental validations.
Another emerging application is the design of multi-parametric biosensors capable of simultaneously monitoring multiple biochemical events. Such sophisticated designs present greater computational challenges but offer unprecedented insights into cellular signaling networks. MD simulations will be crucial for ensuring that these complex biosensors maintain specificity and minimal crosstalk between sensing elements.
As computational power increases and algorithms become more refined, the role of in silico design in biosensor development will continue to expand. The integration of MD simulations with other computational approaches, such as molecular docking and free energy calculations, will provide even more comprehensive predictions of biosensor behavior, further accelerating the development of these powerful molecular tools for biological research and clinical applications.
Conclusion
Genetically encoded FRET biosensors have revolutionized our ability to visualize and quantify biochemical activities with high spatiotemporal resolution in living systems. The continued refinement of biosensor design—through spectral expansion, component optimization, and robust calibration—is breaking down barriers to multiplexing and long-term, quantitative imaging. Future directions point toward the development of even more sophisticated biosensors, including those with near-infrared capabilities for deeper tissue imaging, enhanced modularity for targeting diverse analytes, and integration with emerging technologies like artificial intelligence for data analysis. These advancements promise to unlock deeper insights into complex signaling networks, accelerate drug discovery pipelines, and pave the way for novel clinical diagnostics, ultimately bridging fundamental biological research and therapeutic applications.
References
- 1. Understanding FRET as a Research Tool for Cellular Studies - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC4424985/]
- 2. resonance Förster - Wikipedia energy transfer [https://en.wikipedia.org/wiki/F%C3%B6rster_resonance_energy_transfer]
- 3. Based FRET : Principle Biosensor Recent Advances and... Applications [https://pmc.ncbi.nlm.nih.gov/articles/PMC10136898/]
- 4. Frontiers | Application of FRET in Mechanobiology and... Biosensors [https://www.frontiersin.org/journals/bioengineering-and-biotechnology/articles/10.3389/fbioe.2020.595497/full]
- 5. Advanced FRET normalization allows quantitative analysis of protein interactions including stoichiometries and relative affinities in living cells | Scientific Reports [https://www.nature.com/articles/s41598-019-44650-0]
- 6. QuanTI-FRET: a framework for quantitative FRET measurements in living cells | Scientific Reports [https://www.nature.com/articles/s41598-020-62924-w]
- 7. FRET or No FRET: A Quantitative Comparison - PMC [https://www.ncbi.nlm.nih.gov/pmc/articles/PMC1302980/]
- 8. Expanding Horizons in Advancements of FRET Biosensing Technologies [https://www.mdpi.com/2079-6374/15/7/452]
- 9. Expanding Horizons in Advancements of FRET Biosensing Technologies - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC12293601/]
- 10. Recent advances in FRET-Based biosensors for biomedical applications - ScienceDirect [https://www.sciencedirect.com/science/article/abs/pii/S0003269721002244]
- 11. Review of FRET biosensing and its application in biomolecular detection - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC10006758/]
- 12. Addgene: Fluorescent Guide: Protein Biosensors [https://www.addgene.org/fluorescent-proteins/biosensors/]
- 13. ZEISS Microscopy Online Campus | Interactive Tutorials | Fluorescent ... [https://zeiss-campus.magnet.fsu.edu/tutorials/spectralimaging/fretbiosensors/indexflash.html]
- 14. Developments in FRET - and BRET-Based Biosensors [https://encyclopedia.pub/entry/39777]
- 15. Frontiers | Recent Advances in Biosensor Technology for Potential Applications – An Overview [https://www.frontiersin.org/journals/bioengineering-and-biotechnology/articles/10.3389/fbioe.2016.00011/full]
- 16. Expanding Horizons in Advancements of FRET Biosensing Technologies [https://pubmed.ncbi.nlm.nih.gov/40710102/]
- 17. A Guide to Fluorescent Protein FRET - PMC Pairs [https://pmc.ncbi.nlm.nih.gov/articles/PMC5038762/]
- 18. Characterization of a spectrally diverse set of fluorescent proteins as... [https://www.nature.com/articles/s41598-017-12212-x?error=cookies_not_supported&code=ebaae877-97cc-4798-a794-7f83caf8fdfc]
- 19. How do you choose FRET pairs? | AAT Bioquest [https://www.aatbio.com/resources/faq-frequently-asked-questions/How-do-you-choose-FRET-pairs]
- 20. Mastering FRET Techniques [https://www.numberanalytics.com/blog/mastering-fret-techniques-in-biophysics]
- 21. Fluorescence resonance energy transfer | Abcam [https://www.abcam.com/en-us/technical-resources/guides/fluorescence-guide/fluorescence-resonance-energy-transfer]
- 22. Detection of Protein Interactions in the Cytoplasm and Periplasm of... [https://bio-protocol.org/en/bpdetail?id=2697&type=0]
- 23. Genetically Encoded Fluorescent Biosensors for Biomedical Applications - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC8615007/]
- 24. Genetically encoded fluorescent biosensors illuminate kinase signaling in cancer - PMC [https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6779441/]
- 25. FRET Biosensors for Cancer Detection and Evaluation of Drug Efficacy - PMC [https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2913312/]
- 26. - Real visualization of STAT activation in time using... live cells [https://www.nature.com/articles/s41589-025-02012-0?error=cookies_not_supported&code=5305e581-699b-4321-ae53-6e446ceb8a86]
- 27. FRET Based Biosensor: Principle Applications Recent Advances and Challenges [https://www.mdpi.com/2075-4418/13/8/1375]
- 28. Förster Resonance Energy Transfer ( FRET )-Based Biosensor [https://encyclopedia.pub/entry/43139]
- 29. cAMP Biosensors Based on Genetically Encoded Fluorescent/Luminescent Proteins - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC7911721/]
- 30. Multiplexed Dark FRET : An accessible Biosensors - live platform... cell [https://www.researchsquare.com/article/rs-6580769/v1]
- 31. Genetically Encoded Biosensors Based on Fluorescent Proteins [https://www.mdpi.com/1424-8220/21/3/795]
- 32. Genetically encoded FRET-based biosensors for multiparameter fluorescence imaging - ScienceDirect [https://www.sciencedirect.com/science/article/abs/pii/S0958166909000056]
- 33. - Red for High-Throughput Fluorescence... Shifted FRET Biosensors [https://www.mdpi.com/2079-6374/8/4/99]
- 34. Real-time visualization of small GTPase interactions [https://www.news-medical.net/health/Real-time-visualization-of-small-GTPase-interactions-with-near-infrared-FRET-biosensors.aspx]
- 35. A general method for the development of multicolor biosensors with large dynamic ranges | Nature Chemical Biology [https://www.nature.com/articles/s41589-023-01350-1]
- 36. Two novel red- FRET bosensors in the 670-720 nm range ERK [https://jbioleng.biomedcentral.com/articles/10.1186/s13036-025-00541-9]
- 37. Decoding Molecular Network Dynamics in Cells: Advances in Multiplexed Live Imaging of Fluorescent Biosensors [https://www.mdpi.com/2079-6374/15/9/614]
- 38. Signaling diversity enabled by Rap1-regulated plasma membrane ERK ... [https://elifesciences.org/articles/57410]
- 39. Shedding light on developmental ERK signaling with genetically encoded biosensors - PubMed [https://pubmed.ncbi.nlm.nih.gov/34338283/]
- 40. Development of an optimized backbone of FRET biosensors for kinases and GTPases - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC3226481/]
- 41. Development of a FRET biosensor with high specificity for Akt - PubMed [https://pubmed.ncbi.nlm.nih.gov/24212374/]
- 42. Integration of FRET and sequencing to engineer kinase biosensors ... [https://www.nature.com/articles/s41467-021-25323-x?error=cookies_not_supported]
- 43. Two Novel Red- FRET in the 670–720nm Range - PMC ERK Biosensors [https://pmc.ncbi.nlm.nih.gov/articles/PMC11642818/]
- 44. of Detection - SARS - CoV S 2 on protein between carbon... based FRET [https://pmc.ncbi.nlm.nih.gov/articles/PMC10687278/]
- 45. Peptide substrate screening for the diagnosis of SARS - CoV - 2 using... [https://link.springer.com/article/10.1007/s00604-021-04766-5]
- 46. Application of a Primer Passivated CdTe Quantum Dots as... | CoLab [https://colab.ws/articles/10.1016%2Fj.colsurfa.2025.136152]
- 47. Nucleic Acid Sandwich Hybridization Assay with Quantum Dot-Induced Fluorescence Resonance Energy Transfer for Pathogen Detection - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC3571803/]
- 48. Recent Advances in Fluorescence Resonance Energy Transfer (FRET) Biosensors for Exosomes [https://www.mdpi.com/1467-3045/47/4/235]
- 49. -Based FRET : Genetically Encoded Tools to... | IntechOpen Biosensors [https://www.intechopen.com/chapters/57178]
- 50. FRET Based Biosensor: Principle Applications Recent Advances and Challenges - PubMed [https://pubmed.ncbi.nlm.nih.gov/37189476/]
- 51. Genetically encoded biosensor for fluorescence of... lifetime imaging [https://www.nature.com/articles/s41592-025-02610-9?error=cookies_not_supported&code=080f07bc-3f11-49dd-a764-90a479a1e7e0]
- 52. Opens A New Era in Neuroscience PTEN In Vivo Imaging [https://www.creative-biogene.com/blog/pten-in-vivo-imaging-opens-a-new-era-in-neuroscience]
- 53. Increase your recognition in the scientific world with short video-casts [https://sciencecast.org/casts/fakumgvqoixz]
- 54. Genetically encoded biosensor for fluorescence of... lifetime imaging [https://www.addgene.org/browse/article/28251914/]
- 55. Next-generation Genetically Encoded Fluorescent Biosensors Illuminate Cell Signaling and Metabolism - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC11786609/]
- 56. of Fluorescence- and Bioluminescence-Based... Application [https://pubmed.ncbi.nlm.nih.gov/39727835/]
- 57. real-time imaging of intracellular Quantitative ... FRET biosensor [https://www.nature.com/articles/s41598-020-61478-1?error=cookies_not_supported&code=9517f41f-85c3-476e-818d-ce811c43b93a]
- 58. Förster resonance energy transfer biosensors for fluorescence and... [https://www.nature.com/articles/s41598-022-09364-w?error=cookies_not_supported&code=746ebd00-858e-492a-8d3a-23712e4b4c68]
- 59. MCB | Engineering Zap70 Biosensor Through Directed Evolution for... [https://www.techscience.com/mcb/v16nSuppl.2/35208]
- 60. Frontiers | Resource for FRET-Based Biosensor Optimization [https://www.frontiersin.org/journals/cell-and-developmental-biology/articles/10.3389/fcell.2022.885394/full]
- 61. Comprehensive Guide to Fluorescent ... / BOC Sciences Proteins [https://probes.bocsci.com/resources/comprehensive-guide-to-fluorescent-proteins-and-their-uses-in-research.html]
- 62. Introduction to Fluorescent | Learn... | Leica Microsystems Proteins [https://www.leica-microsystems.com/science-lab/life-science/fluorescent-proteins-introduction-and-photo-spectral-characteristics/]
- 63. Fluorescent protein biosensors [https://www.nature.com/collections/ddbafdbfdj]
- 64. Fluorescent protein biosensors applied to microphysiological systems - PMC [https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4464952/]
- 65. and photostable yellow Bright for extended... fluorescent proteins [https://www.nature.com/articles/s41467-025-58223-5?error=cookies_not_supported&code=10f9b4d9-2591-4246-b77c-218f0adaf984]
- 66. Improving brightness and photostability of green and red... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4754705/]
- 67. Protein engineering and electrochemical biosensors - PubMed [https://pubmed.ncbi.nlm.nih.gov/17960341/]
- 68. An Improved Ratiometric FRET Biosensor with Higher Affinity for Extracellular ATP [https://www.mdpi.com/1424-8220/25/18/5903]
- 69. Engineering FRET for H3K9 acetylation imaging in single... biosensor [https://link.springer.com/article/10.1007/s44258-024-00032-4]
- 70. Directed Evolution to Engineer Monobody for FRET Biosensor Assembly and Imaging at Live-Cell Surface - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC5910193/]
- 71. Dynamic tuning of FRET in a green fluorescent protein biosensor - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC6685724/]
- 72. Fluorescent Proteins as Genetically Encoded FRET Biosensors in Life Sciences [https://www.mdpi.com/1424-8220/15/10/26281]
- 73. Superior robustness of ExEm-spFRET to IIem-spFRET method in... [https://pubmed.ncbi.nlm.nih.gov/30338530/]
- 74. A general method for the development of multicolor biosensors with... [https://www.nature.com/articles/s41589-023-01350-1?error=cookies_not_supported&code=af07a8dd-172e-4ac1-9a86-39e2434ba5d7]
- 75. Two Decades of Genetically Encoded Biosensors Based on Förster Resonance Energy Transfer [https://www.jstage.jst.go.jp/article/csf/44/2/44_18035/_html/-char/en]
- 76. HTRF technology on Microplate Readers | BMG LABTECH [https://www.bmglabtech.com/en/blog/htrf/]
- 77. Dynamic Luminescent Biosensors Based on Peptides for... | IntechOpen [https://www.intechopen.com/chapters/65394]
- 78. A New Generation of FRET Sensors for Robust... | PLOS One [https://journals.plos.org/plosone/article?id=10.1371/journal.pone.0146789]
- 79. ZEISS Microscopy Online Campus | FRET Microscopy with Spectral Imaging [https://zeiss-campus.magnet.fsu.edu/articles/spectralimaging/spectralfret.html]
- 80. An Improved Ratiometric FRET Biosensor with Higher Affinity for Extracellular ATP - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC12473246/]
- 81. Development of High-Performance Whole Cell Biosensors Aided by Statistical Modeling - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC7146887/]
- 82. A platform of BRET- FRET hybrid biosensors for optogenetics... [https://www.nature.com/articles/s41598-018-27174-x?error=cookies_not_supported]
- 83. Advancements in a FRET for Live-Cell... Biosensor [https://pmc.ncbi.nlm.nih.gov/articles/PMC10338669/]
- 84. Fluctuation-based imaging of nuclear Rac1 activation by protein oligomerisation - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC3936235/]
- 85. A FRET reveals spatiotemporal biosensor and functions of... activation [https://www.nature.com/articles/ncomms12674?error=cookies_not_supported&code=d123f5f9-1207-49cf-bf52-3a4d72c1f374]
- 86. -based kinase FRET screen for ERK and AKT... biosensor inhibitor [https://pubmed.ncbi.nlm.nih.gov/31536726/]
- 87. A FRET-based biosensor for the quantification of glucose in culture supernatants of mL scale microbial cultivations | Microbial Cell Factories | Full Text [https://microbialcellfactories.biomedcentral.com/articles/10.1186/s12934-019-1193-y]
- 88. Addgene: cpFRET - FRET based biosensors Kit [https://www.addgene.org/kits/pertz-cpfret-biosensors/]
- 89. Basics of FRET Microscopy | Nikon’s MicroscopyU [https://www.microscopyu.com/applications/fret/basics-of-fret-microscopy]
- 90. Förster resonance energy transfer biosensors for fluorescence and... [https://www.nature.com/articles/s41598-022-09364-w?error=cookies_not_supported&code=6b8c3469-0148-4866-8ddc-a4153a29364b]
- 91. Fluorescence resonance energy transfer ( FRET )-based biosensors ... [https://pubmed.ncbi.nlm.nih.gov/23053099/]
- 92. Quantitative Ratiometric Imaging of FRET-Biosensors in Living Cells - PMC [https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3789067/]
- 93. An intramolecular FRET for the detection of SARS-CoV-2 in... biosensor [https://pubmed.ncbi.nlm.nih.gov/40091661/]
- 94. Turkish Computational and Theoretical Chemistry » Submission » In... [https://dergipark.org.tr/en/pub/tcandtc/issue/91764/1617260]