Strategies for Molecularly Imprinted Polymers with Reduced Non-Specific Binding: From Synthesis to Application
This article provides a comprehensive overview of advanced strategies to minimize non-specific binding in molecularly imprinted polymers (MIPs), a critical challenge limiting their efficacy in analytical and biomedical applications.
Strategies for Molecularly Imprinted Polymers with Reduced Non-Specific Binding: From Synthesis to Application
Abstract
This article provides a comprehensive overview of advanced strategies to minimize non-specific binding in molecularly imprinted polymers (MIPs), a critical challenge limiting their efficacy in analytical and biomedical applications. Tailored for researchers and drug development professionals, it explores the fundamental mechanisms behind non-selective adsorption and details innovative synthesis protocols, including surface modification with surfactants and solid-phase imprinting. The content covers rigorous characterization methods for validating performance, comparative analyses of support materials, and practical troubleshooting for optimizing MIP design. By integrating recent advances in computational modeling and material science, this review serves as a foundational guide for developing high-fidelity MIPs with enhanced selectivity for use in sensitive detection systems and targeted drug delivery.
Understanding and Characterizing Non-Specific Binding in MIPs
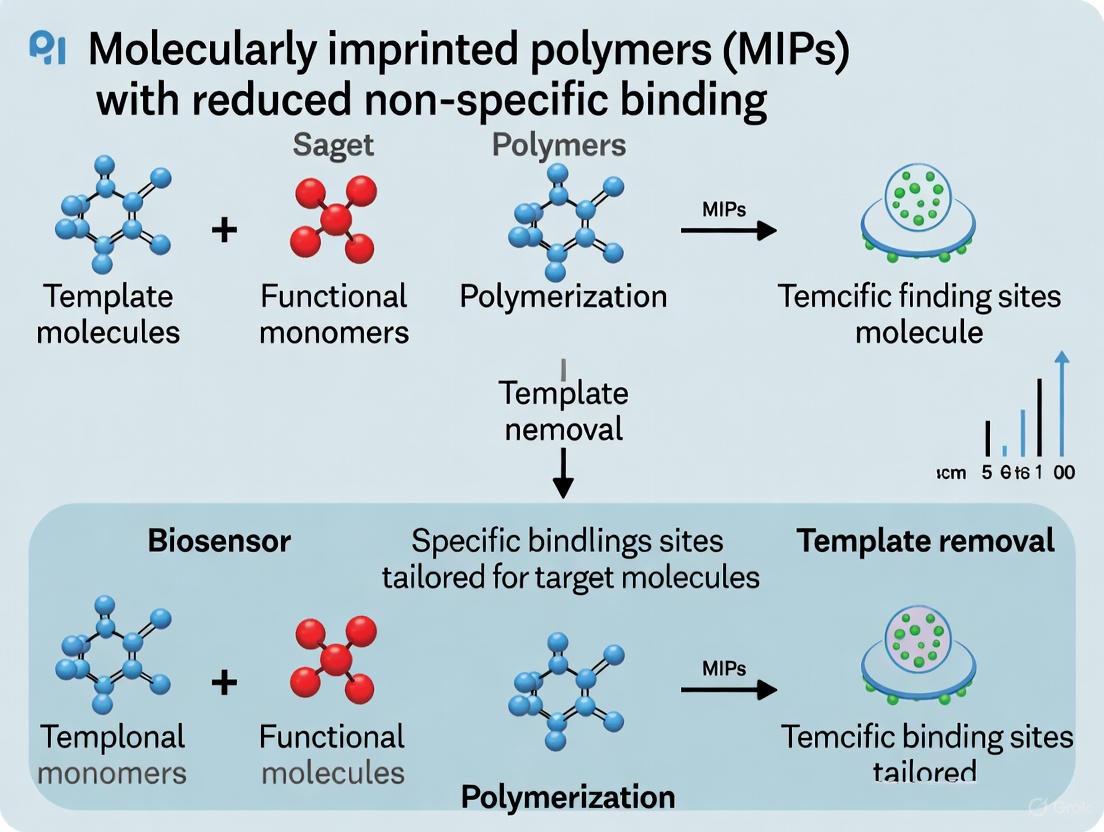
Molecularly imprinted polymers (MIPs) are synthetic biomimetic receptors engineered to exhibit selective binding behavior toward target molecules, functioning as "plastic antibodies" in diagnostic and analytical applications [1]. The fundamental promise of MIP technology lies in creating specific recognition cavities complementary to the template molecule in shape, size, and chemical functionality. However, this promise is compromised by non-specific binding, a phenomenon wherein molecules other than the intended target adhere to non-imprinted regions of the polymer matrix [1]. This non-specific adsorption occurs primarily through interactions with functional groups located outside the meticulously crafted imprinted cavities, significantly reducing the binding specificity and analytical accuracy of MIP-based systems [1] [2]. For researchers and drug development professionals working to translate MIP technology from proof-of-concept to clinical applications, addressing this challenge is paramount for achieving reliable performance in complex biological matrices.
Mechanistic Insights: Origins and Impact of Non-Specific Binding
Structural Heterogeneity in Binding Sites
The molecular architecture of MIPs encompasses both specific binding cavities created during the imprinting process and non-specific sites distributed throughout the polymer matrix. The specific recognition sites result from molecular memory effects, where template molecules are surrounded by functional monomers during polymerization and subsequently extracted, leaving behind complementary cavities [1]. These sites provide the desired selective binding through a combination of shape complementarity and specific chemical interactions such as hydrogen bonding, ionic interactions, and van der Waals forces.
Conversely, non-specific binding originates from:
- Residual functional monomers that did not participate in specific cavity formation
- Chemical heterogeneity in the polymer backbone
- Hydrophobic interactions with the polymer surface
- Electrostatic interactions with charged groups outside imprinted cavities [1]
This structural duality creates a fundamental selectivity challenge, as even optimally imprinted polymers contain a distribution of binding sites with varying affinities and specificities [3].
Analytical Consequences in Sensing Applications
The practical impact of non-specific binding becomes most evident in MIP-based sensors, where false positive signals directly compromise analytical utility. In electrochemical sensors, non-specific adsorption of interferents generates background current that diminishes the signal-to-noise ratio and increases the limit of detection [2]. For optical sensors, non-specific binding can produce false fluorescence or absorbance signals that mask specific binding events. The problem intensifies in complex sample matrices like blood, urine, or environmental samples, where numerous structurally similar compounds may interact non-specifically with the polymer surface [1] [2].
Table 1: Quantitative Comparison of MIP Performance with and without Non-Specific Binding Mitigation
| Parameter | Standard MIP | Surfactant-Modified MIP | Improvement Factor |
|---|---|---|---|
| Binding specificity | 60-75% | >90% | 1.3-1.5x |
| Limit of detection | 15-25 ng mL⁻¹ | 6 ng mL⁻¹ | 2.5-4x |
| Signal-to-noise ratio | Baseline | 2.8-5.1x improvement | Significant enhancement |
| Cross-reactivity | High with structural analogs | Minimal with structural analogs | >70% reduction |
Experimental Strategies to Suppress Non-Specific Binding
Surfactant-Mediated Surface Modification
Electrostatic modification of MIP surfaces with charged surfactants represents a particularly effective approach for minimizing non-specific interactions. This methodology employs surfactant molecules that interact with and effectively block functional groups outside the imprinted cavities while preserving the specific binding sites within the cavities.
Protocol 3.1.1: Surfactant Modification of Conductive Polymer-Based MIPs
Reagents and Materials:
- Synthesized MIP (e.g., polyaniline or polypyrrole-based)
- Sodium dodecyl sulfate (SDS) for positively charged MIP surfaces
- Cetyl trimethyl ammonium bromide (CTAB) for negatively charged MIP surfaces
- Appropriate solvent (e.g., deionized water, buffer solution)
Procedure:
- Prepare a 10 mM solution of the selected surfactant in deionized water
- Immerse the MIP sensor in the surfactant solution for 30 minutes at room temperature with gentle agitation
- Remove the MIP from the surfactant solution and rinse thoroughly with deionized water
- Dry the modified MIP under a stream of nitrogen gas
- Validate modification efficiency through binding assays with the target analyte and potential interferents [1]
This protocol successfully demonstrated complete elimination of non-specific adsorption in MIPs designed for sulfamethoxazole detection, while maintaining high affinity for the target molecule [1].
Protocol 3.1.2: Optimization of Non-Conductive MIPs
For non-conductive polymers such as polydopamine and poly(o-phenylenediamine), an alternative optimization strategy focuses on controlling polymer thickness during electrosynthesis:
- Prepare monomer solution containing the template molecule
- Deposit the polymer film on the electrode surface via cyclic voltammetry
- Systematically vary the number of scans (typically 5-20 cycles) during electrosynthesis
- Evaluate binding specificity after each condition to determine the optimal scan number
- Select the condition that maximizes specific binding while minimizing non-specific adsorption [2]
This approach capitalizes on the inherent properties of non-conductive polymers, where the analyte interacts exclusively through the imprinted cavities rather than the non-conductive polymer matrix itself.
Computational Design and Rational MIP Engineering
Advanced computational approaches now enable rational design of MIPs with minimized non-specific binding potential from the initial synthesis stage. Molecular dynamics (MD) simulations and quantum chemical (QC) calculations help optimize the pre-polymerization mixture to enhance specific interactions while reducing non-specific site formation.
Protocol 3.2.1: Computational Screening of Functional Monomers
- System Setup: Create molecular models of the template and candidate functional monomers (e.g., methacrylic acid, 4-vinylpyridine, acrylamide derivatives)
- Interaction Analysis: Perform QC calculations to determine binding energies and interaction geometries between template and monomers
- Complex Optimization: Identify the most stable template-monomer complexes and their stoichiometric ratios
- Dynamic Simulation: Conduct MD simulations of the pre-polymerization mixture to analyze formation dynamics and stability of template-monomer complexes
- Parameter Definition: Calculate quantitative parameters such as Effective Binding Number (EBN) and Maximum Hydrogen Bond Number (HBNMax) to predict imprinting efficiency [4]
This computational protocol identified that only two molecules of methacrylic acid monomers effectively bind to one molecule of sulfadimethoxine, even when the functional monomer was present in excess (up to 10:1 ratio) [4]. This precise stoichiometric guidance prevents excess functional monomers that would contribute to non-specific binding.
Table 2: Research Reagent Solutions for MIP Development with Reduced Non-Specific Binding
| Reagent Category | Specific Examples | Function in MIP Development | Role in Reducing Non-Specific Binding |
|---|---|---|---|
| Functional Monomers | Methacrylic acid (MAA), 4-vinylpyridine, acrylamide | Form specific interactions with template | Optimal stoichiometry ensures complete participation in specific cavity formation |
| Cross-linkers | EGDMA, TRIM, DVB | Provide structural rigidity to polymer matrix | High cross-linking density preserves cavity integrity and reduces polymer flexibility |
| Surfactant Modifiers | SDS, CTAB | Block non-specific binding sites | Electrostatically neutralize functional groups outside imprinted cavities |
| Computational Tools | MD simulations, QC calculations | Predict optimal synthesis parameters | Guide rational design to maximize specific site formation |
Implementation Workflow: Integrated Strategy for Specificity Enhancement
The following workflow diagram illustrates a comprehensive approach to addressing non-specific binding throughout the MIP development process:
Diagram 1: Comprehensive Workflow for Developing High-Specificity MIPs
This integrated methodology combines computational prediction, rational synthesis, and strategic modification to systematically address non-specific binding at multiple stages of MIP development.
Non-specific binding remains the primary hurdle to achieving optimal specificity in molecularly imprinted polymers, but systematic approaches now exist to effectively mitigate this limitation. The strategic integration of surfactant modification, computational design, and polymer optimization enables researchers to develop MIPs with significantly improved specificity profiles. These advances are particularly crucial for drug development applications where accurate biomarker detection in complex biological matrices is essential. As MIP technology continues to evolve toward clinical implementation, addressing non-specific binding through these multifaceted strategies will be fundamental to achieving the reliability and accuracy required for diagnostic and therapeutic monitoring applications.
Molecularly imprinted polymers (MIPs) are synthetic biomimetic receptors with predetermined selectivity for target analytes, making them ideal for applications in chemical sensing, separation science, and drug delivery [5] [6]. The analytical performance of MIPs is governed by the specific molecular recognition events occurring within tailor-made cavities. However, the practical utility of these polymers is often compromised by two fundamental sources of interference: the chemical nature of external functional groups on the polymer surface and the structural heterogeneity of the imprinted cavities [7] [5]. Non-specific binding (NSB) arising from these factors can significantly inflate analytical signals, reduce selectivity, and lead to erroneous quantitative data [8]. This Application Note delineates the mechanisms of these interference phenomena and provides detailed, actionable protocols for their characterization and mitigation, forming a crucial component of a broader thesis on developing high-fidelity MIPs with minimal non-specific binding.
Mechanisms of Interference
The Role of External Functional Groups
The non-specific adsorption of interfering species onto the MIP's surface is primarily driven by the chemical character of its external functional groups. NSB occurs due to molecular forces between the sample analyte and non-target areas on the polymer surface, including hydrophobic interactions, hydrogen bonding, Van der Waals forces, and electrostatic interactions [7] [8].
- Charged Functional Groups: MIPs synthesized with charged monomers can exhibit strong Coulombic interactions with oppositely charged constituents in the sample matrix. For instance, the overall negative charge of nucleotide-based aptamers makes them prone to non-specific adsorption of positively charged interferents [7]. Similarly, in MIPs, electrostatic NSB can be a significant issue unless carefully managed with uncharged functional monomers [7].
- Hydrophobic Functional Groups: Hydrophobic surfaces can preferentially adsorb hydrophobic protein domains or other non-polar molecules from complex samples like serum or plasma. This is a common challenge in the analysis of biological fluids [8].
The diagram below illustrates the primary mechanisms through which external functional groups contribute to non-specific binding.
The Impact of Cavity Heterogeneity
A defining challenge in MIP synthesis is the formation of a heterogeneous population of binding sites, a direct consequence of the statistical nature of the polymerization process [7]. This heterogeneity manifests as binding sites with varying affinity and specificity for the target molecule.
- High vs. Low-Affinity Sites: Non-covalent imprinting typically yields a polymer characterized by a heterogeneous distribution of binding sites. Scatchard analysis often reveals two distinct classes of sites: a minority of high-affinity, specific sites and a majority of low-affinity, non-specific sites [5]. The latter are a primary source of interference, as they may bind to structural analogs of the template or other matrix components.
- Structural Imperfections: Cavity heterogeneity arises from incomplete complex formation during pre-polymerization, template mobility during polymerization, or damage during template extraction. This results in cavities that are not perfectly complementary to the target in terms of size, shape, and functional group orientation [7] [5]. These imperfect sites lack the selectivity required for specific recognition.
- Macromolecular Imprinting Challenges: For protein templates, the problem is magnified. The large size, structural complexity, and conformational flexibility of proteins make it difficult to create homogeneous, high-fidelity binding sites. Surface imprinting strategies have been developed to address the issue of proteins becoming deeply entrapped in the polymer network, which hinders both template removal and analyte rebinding [7].
The following diagram outlines the origins and consequences of cavity heterogeneity in MIPs.
Table 1: Characterization of MIP Binding Site Heterogeneity
| Binding Site Type | Origin in Polymerization | Affinity Constant (K~d~) | Contribution to Specificity | Contribution to NSB |
|---|---|---|---|---|
| High-Affinity Sites | Optimal template-monomer complex formation | Picomolar (pM) to nanomolar (nM) | High | Negligible |
| Medium-Affinity Sites | Partial complex formation or minor imperfections | Nanomolar (nM) to micromolar (μM) | Moderate | Low to Moderate |
| Low-Affinity/Non-Specific Sites | Random monomer arrangement; no true imprinting | Micromolar (μM) and above | None | High |
Experimental Protocols for Characterization & Mitigation
Protocol 1: Comprehensive Binding Characterization via Batch Rebinding and Scatchard Analysis
This protocol is essential for quantifying the heterogeneity of MIP binding sites and understanding the affinity distribution, which directly relates to non-specific binding potential [5].
1. Materials and Reagents:
- Purified and finely ground MIP particles
- Control Non-Imprinted Polymer (NIP) particles
- Target analyte (high-purity)
- Radiolabeled or fluorescently labeled analyte (for sensitive detection)
- Appropriate buffer (e.g., phosphate-buffered saline, PBS)
- Vacuum filtration setup or centrifugation equipment
- Analytical instrument (e.g., HPLC, LC-MS/MS, scintillation counter, fluorimeter)
2. Procedure:
- Step 1: Preparation of MIP/NIP Suspensions. Precisely weigh 5.0 mg of MIP and NIP into separate glass vials. Add 10 mL of buffer to each vial to create a 0.5 mg/mL suspension. Sonicate for 5 minutes to ensure complete dispersion.
- Step 2: Analyte Spiking. Prepare a stock solution of the target analyte. Spike the MIP and NIP suspensions with a series of analyte concentrations (e.g., 0.1, 0.5, 1, 5, 10, 50, 100 μM). Perform each concentration in triplicate. Include blank samples (polymer without analyte) and control samples (analyte without polymer).
- Step 3: Binding Equilibrium. Cap the vials and incubate on a mechanical shaker for 12-24 hours at a constant temperature (e.g., 25°C) to ensure binding equilibrium is reached.
- Step 4: Separation of Free Analyte. Separate the polymer particles from the solution by vacuum filtration through a 0.22 μm membrane or by high-speed centrifugation (e.g., 15,000 rpm for 10 minutes).
- Step 5: Quantification of Free Analyte. Carefully collect the supernatant and quantify the concentration of the unbound (free) analyte using a calibrated analytical method (HPLC, LC-MS/MS, etc.).
- Step 6: Data Calculation. Calculate the amount of bound analyte (B) for each initial concentration using the equation:
B = (C~i~ - C~f~) * V / m, where C~i~ is the initial concentration, C~f~ is the final free concentration, V is the solution volume, and m is the mass of the polymer. - Step 7: Scatchard Analysis. Plot
B/FversusB, where F is the free analyte concentration at equilibrium. A non-linear Scatchard plot indicates site heterogeneity. Fit the data to a model (e.g., a two-site model) to estimate the dissociation constants (K~d1~, K~d2~) and binding site capacities (N~max1~, N~max2~) for the high- and low-affinity populations [5].
Protocol 2: Mitigation of NSB via Surface Blocking and Buffer Optimization
This protocol describes strategies to minimize NSB on the external surface of MIPs, particularly when used in sensor platforms or solid-phase extraction [8].
1. Materials and Reagents:
- MIP-coated sensor chip, microplates, or MIP particles
- Running buffer (e.g., HEPES, PBS)
- Bovine Serum Albumin (BSA), fraction V
- Non-ionic surfactant (e.g., Tween 20)
- Sodium chloride (NaCl)
- Target analyte and potential interferents
2. Procedure:
- Step 1: pH Optimization. Determine the isoelectric point (pI) of your target analyte. Prepare running buffers at pH values at or near the pI to neutralize the analyte's net charge, thereby reducing charge-based NSB. A common starting range is pH 7.0-7.4 for physiological conditions.
- Step 2: Protein Blocking. Prepare a running buffer containing 0.1% to 1% (w/v) BSA. BSA acts as a blocking agent by adsorbing to non-specific sites on the polymer surface and tubing, preventing the adsorption of your target analyte or interferents. Incubate the MIP surface with this buffer for 30-60 minutes prior to the assay.
- Step 3: Surfactant Addition. To disrupt hydrophobic interactions, add a non-ionic surfactant like Tween 20 to the running buffer at a concentration of 0.005% to 0.05% (v/v). Higher concentrations may risk disrupting specific binding or denaturing biomolecules.
- Step 4: Ionic Strength Shielding. For NSB primarily caused by electrostatic interactions, prepare running buffers with increasing concentrations of NaCl (e.g., 50 mM, 150 mM, 200 mM). The ions in the salt shield the charges on the analyte and the polymer surface, reducing non-specific attraction.
- Step 5: NSB Evaluation. For each optimized buffer condition, run the analyte over a non-imprinted polymer (NIP) surface or a bare sensor surface. The response observed on the NIP under optimized conditions is a direct measure of the remaining NSB. This value should be subtracted from the total binding signal obtained on the MIP to quantify specific binding accurately [8].
Table 2: Research Reagent Solutions for NSB Mitigation
| Reagent / Solution | Primary Function | Mechanism of Action | Typical Working Concentration |
|---|---|---|---|
| Bovine Serum Albumin (BSA) | Protein blocking agent | Saturates non-specific hydrophobic and charged surfaces on the polymer and system | 0.1% - 1.0% (w/v) |
| Tween 20 | Non-ionic surfactant | Disrupts hydrophobic interactions by masking hydrophobic surfaces | 0.005% - 0.05% (v/v) |
| Sodium Chloride (NaCl) | Ionic strength modifier | Shields electrostatic forces by generating an ionic double layer | 50 - 300 mM |
| Phosphate Buffered Saline (PBS) | Standard running buffer | Provides physiological pH and ionic strength | 10 mM phosphate, 137 mM NaCl, 2.7 mM KCl, pH 7.4 |
| HEPES Buffer | Alternative running buffer | Good buffering capacity without forming complexes with metal ions | 10 - 50 mM, pH 7.0-7.6 |
Integrated Workflow for MIP Development with Minimal NSB
The following diagram presents a consolidated workflow for developing and validating MIPs with minimal non-specific binding, integrating the concepts and protocols discussed.
The path to realizing the full potential of MIPs in demanding analytical and biomedical applications lies in a fundamental understanding and systematic mitigation of interference mechanisms. The synergistic challenges posed by external functional groups and intrinsic cavity heterogeneity necessitate a rigorous, two-pronged investigative approach. By implementing the detailed characterization protocols—such as Scatchard analysis to deconvolute binding site populations—and employing strategic buffer optimization and surface blocking techniques, researchers can quantitatively assess and significantly reduce non-specific binding. This Application Note provides a foundational framework for the rational design and validation of high-performance MIPs, a critical step towards their successful integration into robust diagnostic, therapeutic, and environmental monitoring platforms.
The development of advanced polymeric materials, particularly Molecularly Imprinted Polymers (MIPs) with reduced non-specific binding, relies profoundly on robust material characterization techniques. Understanding the intricate relationships between polymer structure, morphology, and function is pivotal for researchers and drug development professionals aiming to design highly selective sensing and separation systems. This article details the integrated application of Brunauer-Emmett-Teller (BET) theory, Fourier-Transform Infrared (FT-IR) Spectroscopy, and Scanning Electron Microscopy (SEM) to comprehensively analyze polymer morphology. Within the context of MIP research, these techniques enable the precise evaluation of structural characteristics, surface functionality, and porosity that directly influence binding efficiency and selectivity, thereby facilitating the creation of superior synthetic receptors with minimized non-specific interactions.
Core Characterization Techniques: Principles and Applications
Scanning Electron Microscopy (SEM) for Morphological Analysis
Principle and Relevance: Scanning Electron Microscopy provides high-resolution, three-dimensional-like images of polymer surfaces and internal structures by scanning a focused beam of electrons across the sample and detecting signals such as secondary electrons (SE) and backscattered electrons (BSE) [9]. For MIP characterization, SEM is indispensable for visualizing surface topography, internal structure, the shape and size of imprinted cavities, and the distribution of phases within polymer blends [10].
Key Applications in MIP Research:
- Cavity Structure Verification: SEM reveals the presence of imprinted cavities and their uniformity. Studies on 4-vinylpyridine MIPs for mandelic acid showed that MIP surfaces had cavities and were rougher than non-imprinted polymers (NIPs), which is a direct morphological indicator of successful imprinting [11].
- Fracture Surface Analysis: Examining fracture surfaces of polymers after tensile tests helps identify failure mechanisms and defects, such as micro-cracks or pores, that can impact material performance and binding properties [10].
- Phase Distribution Assessment: For polymer blends, SEM can identify continuous and dispersed phases. The shape, size, and distribution of these phases provide insights into polymer-additive interactions and the final material's mechanical and thermal properties [10].
Table 1: SEM Analysis Information Outputs for Polymers
| Information Type | Description | Relevance to MIP Development |
|---|---|---|
| Surface Topography | 3D visualization of surface features (roughness, patterns, defects) [9]. | Identifies successful cavity formation and surface area available for binding [11]. |
| Morphological Structure | Shape and size of polymer particles, internal structure from cross-sections [10]. | Reveals porosity and overall morphology critical for template diffusion. |
| Compositional Contrast | Differentiation of materials based on atomic number using Backscattered Electrons (BSE) [9]. | Helps verify the uniform distribution of functional monomers or additives within the polymer matrix. |
| Elemental Distribution | Identification and mapping of elements via Energy-Dispersive X-ray (EDX) analysis [10]. | Confirms the presence and dispersion of specific catalytic or functional elements. |
Fourier-Transform Infrared (FT-IR) Spectroscopy for Structural and Functional Group Analysis
Principle and Relevance: FT-IR spectroscopy identifies functional groups and chemical bonds within a polymer by measuring the absorption of infrared light at specific wavelengths [12]. It provides critical information on the chemical structure of the repeat units and can confirm successful polymerization and template-monomer interactions in MIPs.
Key Applications in MIP Research:
- Monitoring Polymerization: FT-IR spectra can confirm the formation of the polymer network by tracking the disappearance of monomer double bonds (C=C) and the appearance of new linkages [11].
- Verifying Template Removal and Rebinding: Spectral shifts or intensity changes in characteristic functional group bands (e.g., C=O, O-H, N-H) can indicate the successful removal of the template molecule and its subsequent rebinding to the imprinted cavities [13] [11]. For instance, in polystyrene/bitumen composites, a shift and constriction of the O-H stretching bands upon bitumen incorporation indicated a strong interaction between the components [13].
- Detecting Undesired Interactions: FT-IR can help identify the chemical basis for non-specific binding, such as the presence of accessible functional groups outside the imprinted cavities that interact non-selectively with analyte molecules [1].
Table 2: Key FT-IR Spectral Interpretations for Polymers
| Functional Group / Vibration | Typical Wavenumber (cm⁻¹) | Interpretation and Significance |
|---|---|---|
| O-H Stretching | 3200-3600 | Indicates presence of alcohols, carboxylic acids; shifts can signal hydrogen bonding with templates [13]. |
| C-H Stretching (CH₂) | ~2917 (asym), ~2852 (sym) | Characteristic of polymer backbones; used to identify polyethylene and similar structures [12]. |
| C=O Stretching | ~1700 | Suggests presence of esters or carboxylic acids from monomers like MAA or cross-linkers like EGDMA. |
| C=C Stretching (Aromatic) | ~1600, ~1500 | Confirms presence of aromatic rings in monomers like styrene or 4-vinylpyridine [11]. |
| C-N Stretching | ~1200-1350 | Can indicate the involvement of amine-containing monomers in binding interactions. |
BET (Brunauer-Emmett-Teller) Theory for Surface Area and Porosity Analysis
Principle and Relevance: The BET theory is the standard method for determining the specific surface area of porous materials by analyzing nitrogen gas adsorption-desorption isotherms at cryogenic temperatures. It also provides information on pore size distribution and total pore volume. For MIPs, a high surface area is often correlated with a greater number of accessible imprinted sites, while pore size dictates the diffusion and accessibility of the target molecule.
Key Applications in MIP Research:
- Optimizing Porogenic Conditions: BET analysis is used to optimize the type and amount of porogen during MIP synthesis, as the porogen directly influences the material's final porous structure [11] [14].
- Correlating Structure with Performance: A direct correlation can be established between the surface area and binding capacity of MIPs. A higher surface area typically provides more binding sites for the target molecule.
- Quality Control: BET provides a quantitative measure to ensure batch-to-batch consistency in MIP synthesis, which is crucial for commercial applications.
Experimental Protocols for MIP Characterization
Protocol: SEM Analysis of Molecularly Imprinted Polymers
Objective: To characterize the surface morphology and internal structure of MIPs and NIPs.
Materials and Equipment:
- Scanning Electron Microscope
- Sputter coater (e.g., for gold or carbon coating)
- Conductive adhesive tape (e.g., carbon tape)
- Sample stubs
- Liquid nitrogen (for cryogenic fracturing, if needed)
Procedure:
- Sample Preparation:
- For surface morphology, mount a small amount of dry MIP or NIP powder directly onto a sample stub using conductive double-sided tape [9].
- For cross-sectional analysis, freeze the polymer in liquid nitrogen and fracture it to expose the internal structure. Mount the fractured piece to reveal the cross-section [10].
- Conductive Coating: Due to the insulating nature of most polymers, coat the mounted samples with a thin (a few nanometers) layer of a conductive material like gold or carbon using a sputter coater. This step prevents charging effects that distort the image [9] [10].
- SEM Imaging:
- Place the coated sample into the SEM chamber.
- Select an appropriate accelerating voltage (typically 5-20 kV for polymers). Lower voltages can help minimize damage to sensitive polymer surfaces.
- Begin with low magnification to locate an area of interest, then increase magnification to visualize morphological details, cavities, and surface texture [9] [10].
- Capture images using both secondary electron (SE) mode for topography and backscattered electron (BSE) mode for compositional contrast.
Data Interpretation: Compare MIP and NIP micrographs. Successful imprinting is often indicated by a rougher surface texture and the presence of pores or cavities in the MIP that are absent in the smoother, more featureless NIP [11].
Protocol: FT-IR Spectroscopy for MIP Characterization
Objective: To confirm chemical structure, monitor template removal, and investigate binding interactions.
Materials and Equipment:
- FT-IR Spectrometer
- Hydraulic press for KBr pellets (if using transmission mode)
- ATR (Attenuated Total Reflectance) accessory (preferred for powders)
Procedure:
- Sample Preparation:
- Transmission Mode: Grind ~1 mg of dry polymer with ~100 mg of dry potassium bromide (KBr). Compress the mixture into a transparent pellet using a hydraulic press.
- ATR Mode: This is the most common and straightforward method. Place a small amount of dry polymer powder directly onto the ATR crystal and clamp it to ensure good contact. No additional preparation is needed.
- Data Acquisition:
- For transmission mode, place the KBr pellet in the spectrometer's sample holder.
- For ATR mode, ensure the sample is in firm contact with the crystal.
- Acquire a background spectrum (empty crystal or pure KBr pellet).
- Collect the sample spectrum over a range of 4000-400 cm⁻¹ with a resolution of 4 cm⁻¹. Average multiple scans (e.g., 32) to improve the signal-to-noise ratio.
- Template Removal Check: Acquire the FT-IR spectrum of the MIP before and after the template extraction process (e.g., via Soxhlet extraction with a solvent like methanol/acetic acid). The spectrum after elution should resemble that of the NIP, indicating successful template removal [11].
Data Interpretation: Analyze the spectra for characteristic functional group bands. A successful imprinting process may be evidenced by slight shifts in the spectra of the MIP before washing compared to the NIP, which then become nearly identical after template elution [11]. Shifts in bands upon rebinding can indicate specific interactions between the template and the functional groups within the cavities.
Research Reagent Solutions for MIP Development
Table 3: Essential Materials for Molecularly Imprinted Polymer Research
| Reagent / Material | Function and Application | Example in Context |
|---|---|---|
| Functional Monomers | Provide functional groups for interaction with the template molecule. | Methacrylic acid (MAA), 4-Vinylpyridine (4-VP) [11] [15]. |
| Cross-linking Agents | Create a rigid polymer network to stabilize the imprinted cavities. | Ethylene glycol dimethacrylate (EGDMA) [11] [15]. |
| Initiators | Generate free radicals to start the polymerization reaction. | Azobisisobutyronitrile (AIBN) [11] [15]. |
| Porogenic Solvents | Dissolve all components and create pores during polymerization. | Acetonitrile (ACN), Toluene, Dimethylformamide (DMF) [11] [15]. |
| Surfactants | Used to modify MIP surfaces to suppress non-specific adsorption. | Sodium dodecyl sulfate (SDS), Cetyl trimethyl ammonium bromide (CTAB) [1] [16]. |
Integrated Workflow for MIP Characterization
The following diagram illustrates the logical sequence of characterization techniques in the development and analysis of Molecularly Imprinted Polymers.
Case Study: Characterizing MIPs for Reduced Non-Specific Binding
Background: A key challenge in MIP technology is non-specific adsorption on functional groups located outside the imprinted cavities, which reduces selectivity and sensing efficacy [1] [16].
Integrated Characterization Approach:
- Problem Identification (FT-IR): FT-IR analysis can identify the presence of excess, unbound functional groups on the polymer surface that are responsible for non-specific interactions.
- Morphological Assessment (SEM): SEM imaging is performed before and after surface modification to ensure that the surfactant treatment does not damage the imprinted cavities or block the porous structure. The rough, cavity-rich morphology of the MIP should be preserved [11].
- Surface Area Verification (BET): BET analysis confirms that the surface modification process does not significantly reduce the surface area or block the pores, which would hinder access to the specific binding sites.
- Solution Implementation: Researchers successfully suppressed non-specific binding by electrostatically modifying MIPs with surfactants like SDS and CTAB. The surfactants react with the external functional groups, "blocking" them from non-specifically binding interferents [1] [16].
- Performance Validation: Post-modification, binding isotherms showed that the modified MIPs retained high adsorption capacity for the target molecule (sulfamethoxazole) while significantly reducing non-specific uptake, as evidenced by the low binding of the corresponding NIP [1].
This multi-technique approach ensures that strategies to reduce non-specific binding effectively enhance selectivity without compromising the structural integrity or specific binding capacity of the MIP.
The Impact of Polymer Synthesis Parameters on Binding Site Fidelity
Within the broader research on developing molecularly imprinted polymers (MIPs) with reduced non-specific binding, controlling the fidelity of the synthesized binding sites is paramount. Binding site fidelity refers to the accuracy with which the imprinted cavities complement the template molecule in size, shape, and chemical functionality. High-fidelity sites are characterized by their high affinity and selectivity, which are critical for applications in sensitive detection, separation, and drug delivery [17]. The synthesis of MIPs involves a complex interplay of components and conditions, including the choice of functional monomer, cross-linker, solvent, and polymerization technique. Any variation in these parameters can significantly impact the heterogeneity of the binding sites, which is the defining characteristic of MIPs, ultimately affecting their performance by introducing non-specific binding [18]. These Application Notes and Protocols provide a detailed quantitative and methodological guide for researchers aiming to systematically optimize these synthesis parameters to achieve high binding site fidelity.
Quantitative Impact of Synthesis Parameters
The tables below summarize the quantitative effects of key synthesis parameters on binding site fidelity, as established by computational and experimental studies.
Table 1: Quantitative Parameters for Monomer-Template Interaction from Computational Chemistry
| Parameter | Description | Impact on Binding Site Fidelity | Experimental Correlation |
|---|---|---|---|
| Binding Energy (ΔEbind) | Energy released upon template-monomer complex formation in vacuum [4]. | Higher negative values indicate more stable pre-polymerization complexes, leading to higher fidelity sites. | A ΔEbind of -82.30 kJ/mol for a double hydrogen-bonded complex vs. -30.17 kJ/mol for a single bond showed significantly improved stability [4]. |
| Effective Binding Number (EBN) | The average number of monomer molecules effectively bound to a single template molecule in the pre-polymerization mixture [4]. | Higher EBN values suggest a more stable and well-defined imprint, leading to higher fidelity. | In a system with a 10:1 monomer-to-template ratio, the EBN was only 2, guiding the optimal synthesis ratio [4]. |
| Maximum H-Bond Number (HBNMax) | The maximum number of hydrogen bonds possible between the template and a functional monomer [4]. | Higher HBNMax contributes to greater complex stability and higher fidelity recognition. | Carboxylic acid monomers formed complexes with double hydrogen bonds (e.g., N-H⋯O=C and S=O⋯H-O), resulting in higher ΔEbind and improved fidelity [4]. |
Table 2: Impact of Polymerization Composition and Conditions on Site Fidelity
| Parameter | Typical Optimal Range | Impact on Binding Site Fidelity | Rationale |
|---|---|---|---|
| Monomer Type | Carboxylic acids (e.g., MAA, TFMAA) | Higher fidelity compared to ester monomers [4]. | Carboxylic acids offer both hydrogen bond donor and acceptor groups, enabling stronger, multi-point interactions with the template [4]. |
| Template : Monomer : Crosslinker | 1 : 3-6 : 30 (e.g., for cortisol MIP) [4] | A balanced ratio is critical; excess monomer can promote non-specific binding. | This ratio maximizes the effective binding efficiency (EBN) while ensuring sufficient cross-linking to stabilize the imprinted cavities [4]. |
| Solvent (Porogen) | Low polarity (e.g., Acetonitrile, Toluene) | Enhances fidelity by promoting template-monomer interactions [17]. | Low-polarity solvents do not compete with the template for hydrogen bonding with the functional monomer, strengthening the pre-polymerization complex [17]. |
| Polymerization Technique | Surface-initiated (e.g., SI-SARA ATRP) [4] | Higher fidelity than bulk polymerization for large templates. | Creates binding sites at the surface, improving template removal and access, which reduces site heterogeneity and non-specific binding [17]. |
Experimental Protocols
Protocol 1: Computational Screening of Functional Monomers
This protocol utilizes molecular dynamics (MD) simulations to define quantitative parameters for monomer selection prior to synthesis, saving time and resources [4].
Methodology:
- System Setup: Model the template molecule and candidate functional monomers (e.g., methacrylic acid, acrylamide, 4-vinylpyridine) using computational chemistry software. Optimize all geometries using quantum chemical methods (e.g., DFT at the B3LYP/6-31G(d) level) [4].
- MD Simulation: Simulate the pre-polymerization system in an explicit solvent box (e.g., acetonitrile). Include the cross-linker (e.g., EGDMA) at the intended synthesis ratio.
- Trajectory Analysis:
- Calculate the Effective Binding Number (EBN) by analyzing the simulation trajectory to determine the average number of monomer molecules bound to the template.
- Calculate the Maximum Hydrogen Bond Number (HBNMax) by identifying all possible hydrogen bond donors and acceptors and their occupancy during the simulation.
- Selection Criterion: Select the functional monomer that yields the highest values of EBN and HBNMax, indicating a higher probability of forming stable, high-fidelity binding sites.
Protocol 2: Solid-Phase Synthesis of High-Affinity nanoMIPs
This protocol describes an advanced imprinting technique for producing MIP nanoparticles with improved binding site uniformity and reduced non-specific binding [19].
Methodology:
- Immobilization: Covalently immobilize the template molecules onto solid beads (e.g., glass or silica).
- Monomer Mixture Incubation: Incubate the template-grafted beads with a solution containing the functional monomer, cross-linker, and initiator.
- Surface-Initiated Polymerization: Initiate polymerization, typically via a controlled radical polymerization method (e.g., SI-SARA ATRP as used in [4]), to grow a thin, cross-linked polymer layer around the template.
- Template Removal and Harvesting: Wash the beads with a strong solvent to remove the template, creating specific cavities. Release the resulting nanoMIPs from the solid support into solution. This method confines imprinting to the surface, creating more homogeneous and accessible binding sites.
Signaling Pathways and Workflows
The following diagram illustrates the logical workflow for rational design of high-fidelity MIPs, integrating computational and experimental approaches.
MIP Rational Design Workflow
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Materials for Rational MIP Development
| Reagent / Material | Function / Role in Enhancing Fidelity | Specific Example(s) |
|---|---|---|
| Carboxylic Acid Monomers | Serve as functional monomers; their dual hydrogen-bonding capability promotes stable complex formation with templates [4]. | Methacrylic acid (MAA), Acrylic acid (AA), Trifluoromethylacrylic acid (TFMAA) [4]. |
| Cross-linkers | Create a rigid polymer network that stabilizes the imprinted cavities, "freezing" them in the correct configuration to prevent collapse and maintain fidelity [17]. | Ethylene glycol dimethacrylate (EGDMA) [4]. |
| Low-Polarity Solvents (Porogens) | The solvent medium for polymerization; low polarity enhances hydrogen bonding between template and monomer, improving pre-polymerization complex stability [17]. | Acetonitrile, Toluene. |
| Controlled Radical Initiators | Enable surface-initiated polymerization techniques (e.g., SI-SARA ATRP), which produce more uniform MIP nanoparticles with better-defined binding sites [4]. | Supplemental Activator and Reducing Agent for ATRP. |
| Computational Software | Used for quantum chemical calculations and MD simulations to predict template-monomer interaction strength and guide monomer selection prior to synthesis [4]. | Gaussian, GROMACS. |
Synthesis Protocols and Material Innovations for Enhanced Specificity
Surface Molecular Imprinting Technique (SMIT) represents a significant advancement over traditional molecular imprinting by confining the creation of recognition sites to the surface of solid substrates. This approach addresses critical limitations of conventional bulk imprinting, including incomplete template removal, slow mass transfer kinetics, and the "embedding" of binding sites within the polymer matrix [20]. In SMIT, the molecular imprinting process occurs exclusively on the surface of solid-phase matrices, resulting in recognition sites distributed on the outer layer of the substrates [20]. This strategic confinement yields polymers with accessible binding cavities, faster mass transfer rates, and reduced template residue compared to their bulk counterparts [20] [21].
The fundamental advantage of surface imprinting lies in its ability to create recognition sites that are readily available for target molecules, rather than buried within a polymer network. This accessibility is particularly crucial for imprinting large biological templates such as proteins, which face challenges in traditional imprinting due to their size, complexity, and sensitivity to organic solvents [21]. The surface imprinting process typically involves three key stages: (1) complex formation between template and functional monomers on the solid substrate surface, (2) polymerization to form a thin imprinting layer containing templates, and (3) template removal to create specific recognition cavities [20]. This methodology has expanded the application scope of molecularly imprinted polymers (MIPs) to include sensors, separation and purification systems, catalytic platforms, and biomedical devices [20].
Theoretical Foundations and Advantages
Comparative Advantages of Surface Imprinting
Surface imprinting technology resolves several intrinsic problems associated with conventional bulk imprinting methods. The accessibility of binding sites in surface-imprinted polymers significantly enhances binding kinetics, as target molecules no longer need to diffuse through a dense polymer network to reach recognition cavities [20] [21]. This advantage is particularly pronounced for large biomolecules such as proteins, which exhibit slow diffusion rates in traditional MIPs [21].
A second critical advantage involves improved template removal. In bulk imprinting, complete extraction of template molecules is often challenging, leading to persistent template leakage (bleeding) that compromises analytical applications [21]. Surface-imprinted polymers facilitate more efficient template removal and reduce the risk of template leakage, as the recognition sites are openly exposed to the extraction solvent [20]. This characteristic is essential for applications requiring high accuracy, such as sensors and diagnostic assays.
The heterogeneity of binding sites presents a third area of improvement. Traditional bulk MIPs typically contain heterogeneous populations of binding sites with varying affinities and specificities, contributing to significant non-specific binding [21]. Surface confinement allows for more uniform binding sites, as the polymerization process can be better controlled in two dimensions rather than three [20] [21]. This homogeneity translates to enhanced selectivity and reduced non-specific interactions, which is crucial for applications in complex matrices like biological fluids or environmental samples [1].
Table: Comparative Analysis of Bulk vs. Surface Imprinting Techniques
| Parameter | Bulk Imprinting | Surface Imprinting |
|---|---|---|
| Binding Site Accessibility | Sites embedded within polymer matrix | Sites confined to surface layer |
| Mass Transfer Kinetics | Slow diffusion through polymer network | Rapid access to surface sites |
| Template Removal | Often incomplete, potential for leakage | Efficient extraction from surface |
| Binding Site Heterogeneity | High heterogeneity, polyclonal character | More uniform, controlled sites |
| Suitability for Large Templates | Poor for proteins and macromolecules | Excellent for biomacromolecules |
| Non-Specific Binding | Significant due to buried functional groups | Reduced through surface engineering |
Recognition Mechanisms and Binding Site Architecture
The molecular recognition mechanism in surface-imprinted polymers involves complementary interactions between the target molecule and the fabricated binding cavities. These interactions include hydrogen bonding, electrostatic interactions, hydrophobic effects, and van der Waals forces, depending on the functional monomers employed [20] [22]. The recognition process depends on both the three-dimensional geometry of the cavity and the spatial arrangement of functional groups within it [20].
In surface imprinting, the binding site architecture can be precisely controlled through the selection of functional monomers, cross-linkers, and the solid substrate properties [20]. The spatial confinement afforded by surface imprinting enables more consistent cavity dimensions and functional group orientation compared to bulk polymers [21]. This control is essential for achieving high selectivity, particularly when distinguishing between structurally similar molecules in complex mixtures.
The solid substrate used in surface imprinting plays a crucial role in determining the properties of the resulting MIP. Common substrates include silica nanoparticles, quantum dots, iron oxide, graphene oxides, and gold nanoparticles [20]. These substrates provide the foundation for imprinting and can contribute additional functionalities such as magnetism, fluorescence, or conductivity to the composite material [20]. The substrate surface chemistry influences the orientation of template molecules during imprinting, thereby affecting the quality and specificity of the resulting binding sites [15].
Surface Imprinting Methodologies
Core Surface Imprinting Strategies
Several specialized approaches have been developed to optimize the surface imprinting process for different applications and template types:
Epitope-Mediated Imprinting offers an efficient alternative to whole-protein imprinting. This method utilizes short peptide sequences (epitopes) characteristic of the target protein as templates [21]. The epitope approach provides relatively easy template removal, generates uniform binding sites, and reduces synthesis costs, especially for expensive protein templates [21]. A significant challenge lies in identifying appropriate linear epitopes that accurately represent the native protein structure [21].
Sacrificial Substrate Imprinting involves immobilizing template molecules onto the surface of a sacrificial material such as SiO₂, which is immersed in the monomer mixture during polymerization [21]. Following polymerization, the sacrificial material is dissolved, leaving behind binding sites occupied by template [21]. This method stabilizes protein structure, expands the range of solvents available for imprinting, prevents protein aggregation, and facilitates mass transfer kinetics [21].
Nanomaterial-Assisted Imprinting combines surface imprinting with nanotechnology by using nanomaterials as sacrificial molds or solid supports [21]. This approach provides precise control over the morphology of the imprinted polymer, creating nanostructured materials in the form of nanorods, nanofilaments, or ordered cavities [21]. The nano-structuring significantly enhances the MIP surface area and consequently improves sensitivity, detectability, and response time in sensor applications [21].
Protocol: Surface Imprinting with Silanized Molds for Enhanced Selectivity
This protocol describes a method for creating surface-imprinted polymers using silanized silica molds to minimize non-specific binding, particularly effective for small molecule targets such as the herbicide 2,4-D [15].
Table: Reagents and Materials for Silanized Mold Imprinting
| Reagent/Material | Function | Specifications/Alternatives |
|---|---|---|
| Silica Colloids | Sacrificial mold for patterning | 500 nm-1 μm diameter, monodisperse |
| Trimethoxy(methyl)silane | Silanizing agent for mold surface | Reduces surface functionality |
| Methacrylic Acid (MAA) | Functional monomer | Provides carboxyl groups for template interaction |
| Ethylene Glydimethacrylate (EGDMA) | Cross-linker | Creates rigid polymer network |
| 2,2'-Azobisisobutyronitrile (AIBN) | Photoinitiator | Decomposes under UV to generate radicals |
| 2,4-Dichlorophenoxyacetic Acid (2,4-D) | Template molecule | Target analyte for imprinting |
| Dimethylformamide (DMF) | Solvent | Dissolves monomer/template complex |
Step 1: Preparation of Silanized Silica Molds
- Create close-packed polystyrene (PS) microspheres on a glass substrate through spin-coating and interface assembly techniques [15].
- Generate a porous hexagonal-patterned PDMS mold using the PS colloidal monolayer as a sacrificial mask.
- Replicate hemispherical SiO₂ films using the patterned PDMS mold as a negative template.
- Treat the silica surface with trimethoxy(methyl)silane to reduce surface hydroxyl groups that contribute to non-specific binding [15].
Step 2: Monomer-Template Complex Formation
- Dissolve the functional monomer (MAA, 0.5 mmol) and template (2,4-D, 0.125 mmol) in DMF (2.5 mL) in a glass vial.
- Allow the mixture to pre-associate for 30 minutes with gentle stirring to facilitate complex formation through hydrogen bonding and electrostatic interactions.
Step 3: Photopolymerization with Silanized Mold
- Add cross-linker (EGDMA, 2.5 mmol) and photoinitiator (AIBN, 20 mg) to the monomer-template solution.
- Degas the mixture with nitrogen for 5 minutes to remove oxygen that inhibits polymerization.
- Place the silanized silica mold in contact with the monomer solution on a support substrate.
- Expose the assembly to UV light (365 nm, 10 mW/cm²) for 10 minutes to initiate polymerization [15].
Step 4: Template Removal and Characterization
- Carefully separate the polymer film from the silanized mold.
- Extract template molecules using a methanol-acetic acid solution (9:1 v/v) until no template is detected in the wash solution by UV spectroscopy.
- Validate the imprinting effect by comparing binding capacity with non-imprinted polymers and calculating the imprinting factor (IF = QMIP/QNIP) [15].
Protocol: Surfactant Modification to Suppress Non-Specific Adsorption
This protocol describes an effective method for reducing non-specific binding in molecularly imprinted polymers through electrostatic modification with surfactants, demonstrated for sulfamethoxazole (SMX) imprinting [1] [16].
Table: Reagents for Surfactant-Modified MIPs
| Reagent | Function | Role in Reducing Non-Specific Binding |
|---|---|---|
| Sodium Dodecyl Sulfate (SDS) | Anionic surfactant | Blocks external functional groups in positively charged MIPs |
| Cetyl Trimethyl Ammonium Bromide (CTAB) | Cationic surfactant | Blocks external functional groups in negatively charged MIPs |
| 4-Vinylpyridine | Basic functional monomer | Forms positively charged polymer matrix |
| Methacrylic Acid | Acidic functional monomer | Forms negatively charged polymer matrix |
| Sulfamethoxazole | Template molecule | Antibiotic target analyte |
Step 1: MIP Synthesis
- For positively charged MIPs: Polymerize 4-vinylpyridine (functional monomer) with ethylene glycol dimethacrylate (cross-linker) in the presence of SMX template [1].
- For negatively charged MIPs: Polymerize methacrylic acid with EGDMA in the presence of SMX template [1].
- Extract templates using appropriate solvents to create specific binding cavities.
Step 2: Surfactant Modification
- Prepare SDS solution (10 mM in deionized water) for modifying positively charged MIPs.
- Prepare CTAB solution (10 mM in deionized water) for modifying negatively charged MIPs.
- Incubate MIP particles with surfactant solution (1:10 w/v) for 2 hours at room temperature with gentle agitation [1].
- Recover modified MIPs by centrifugation and rinse lightly with water to remove excess surfactant.
Step 3: Binding Capacity Assessment
- Incubate surfactant-modified MIPs (MIP±-SDS/CTAB) with SMX solutions of varying concentrations.
- Measure adsorption isotherms and compare with non-imprinted polymers (NIPs) and unmodified MIPs.
- Calculate imprinting factors (IF = QMIP/QNIP) to quantify the improvement in specificity [1].
Key Results: Surfactant modification effectively eliminates non-specific adsorption while preserving specific binding through imprinted cavities. The modified MIPs achieve detection limits as low as 6 ng mL⁻¹ for SMX and maintain stability at high temperatures, making them suitable for on-site applications [1].
Analytical Performance and Applications
Quantitative Performance of Surface-Imprinted Polymers
Surface imprinting techniques have demonstrated remarkable performance across various analytical applications, particularly in sensing and separation. The confinement of recognition sites to accessible surfaces significantly enhances binding kinetics and reduces non-specific interactions, leading to improved sensitivity and selectivity [20] [1].
Table: Performance Metrics of Surface-Imprinted Polymers in Sensing Applications
| Target Analyte | Matrix | Detection Platform | Limit of Detection | Imprinting Factor | Reference |
|---|---|---|---|---|---|
| 2,4-D Herbicide | Water | QCM with silanized mold | - | 3.38 (vs. 1.86 with non-silanized) | [15] |
| Sulfamethoxazole | Milk, Water | Surfactant-modified MIP | 6 ng mL⁻¹ | Significantly improved | [1] |
| L-Thyroxine | Buffer | NanoMIP-based ELISA | 8 pM | >10-fold vs. antibodies | [23] |
| Fumonisin B2 | - | NanoMIP-based ELISA | pM range | Comparable to antibodies | [23] |
| Biotin | - | NanoMIP-based ELISA | pM range | Comparable to antibodies | [23] |
The enhanced performance of surface-imprinted polymers is particularly evident in their application as synthetic antibodies in assay formats. Molecularly imprinted polymer nanoparticles (nanoMIPs) prepared by surface imprinting have demonstrated comparable or superior performance to commercially produced antibodies in enzyme-linked competitive assays [23]. These nanoMIPs showed detection limits in the pM range and maintained stability when stored at room temperature for at least one month, offering significant advantages over biological antibodies that require cold chain logistics [23].
Application in Biosensing Platforms
Surface-imprinted polymers have found extensive application in electrochemical and optical biosensors, where they serve as robust recognition elements. In electrochemical sensors, surface-imprinted layers are deposited directly onto electrode surfaces, where they selectively capture target molecules, resulting in measurable changes in electrical signals [20]. The confined recognition sites in surface-imprinted films facilitate rapid binding kinetics and efficient signal transduction, enabling real-time monitoring of analytes [20] [24].
The integration of surface-imprinted polymers with nanozyme systems has created innovative biosensing platforms that combine molecular recognition with catalytic amplification. In these hybrid systems, the surface-imprinted layer provides specific target recognition, while the nanozyme component generates detectable signals through enzyme-mimetic catalysis [25]. This approach has been successfully applied in colorimetric, fluorescence, and electrochemical assays for detecting drugs, pollutants, and disease biomarkers [25].
Another significant advancement involves the development of theranostic applications for cancer diagnosis and treatment. Surface-imprinted polymers designed to recognize specific cancer biomarkers can simultaneously serve for diagnostic imaging and targeted drug delivery [22]. The target specificity of these materials improves therapeutic efficacy while reducing off-target effects, demonstrating the versatility of surface imprinting in advanced biomedical applications [22].
The Scientist's Toolkit: Essential Research Reagents
Successful implementation of surface imprinting techniques requires careful selection of reagents and materials optimized for specific applications and target molecules.
Table: Essential Research Reagents for Surface Imprinting
| Reagent Category | Specific Examples | Function in Surface Imprinting |
|---|---|---|
| Functional Monomers | Methacrylic acid (MAA), 4-Vinylpyridine, Acrylamide | Provide complementary interactions with template molecules |
| Cross-linkers | Ethylene glycol dimethacrylate (EGDMA), N,N'-Methylenebisacrylamide | Create rigid polymer network around template |
| Solid Substrates | Silica nanoparticles, Magnetic beads, Graphene oxides, Gold surfaces | Provide foundation for surface imprinting |
| Surface Modifiers | Trimethoxy(methyl)silane, (3-Aminopropyl)triethoxysilane | Control surface functionality of substrates and molds |
| Non-Specific Binding Blockers | SDS, CTAB, 2-Methacryloyloxyethyl phosphorylcholine | Suppress non-specific interactions |
| Polymerization Initiators | AIBN, Ammonium persulfate/TEMED | Generate free radicals for polymerization |
Surface imprinting techniques have revolutionized the field of molecularly imprinted polymers by confining recognition sites to accessible surfaces, thereby addressing fundamental limitations of traditional bulk imprinting. The strategic placement of binding cavities on material surfaces enables rapid binding kinetics, efficient template removal, and reduced non-specific interactions - critical advantages for applications in sensing, separation, and biomedical engineering [20] [21].
The continued advancement of surface imprinting methodologies, including epitope-mediated approaches, sacrificial substrate techniques, and nanomaterial-assisted imprinting, continues to expand the capabilities of these synthetic recognition materials [21]. Furthermore, innovative strategies such as surfactant modification and mold silanization provide effective solutions to the persistent challenge of non-specific binding [1] [15]. As these technologies mature, surface-imprinted polymers are poised to play an increasingly significant role in analytical chemistry, biomedical diagnostics, and therapeutic applications, potentially rivaling or surpassing the performance of biological recognition elements in specific contexts [23].
The integration of surface imprinting with emerging nanomaterials and signal transduction mechanisms will likely yield even more sophisticated recognition systems with enhanced sensitivities and specificities. These developments hold particular promise for point-of-care diagnostics, environmental monitoring, and targeted therapeutic delivery, where robust, cost-effective recognition elements are essential [25] [22].
Molecularly imprinted polymers (MIPs) are synthetic materials designed with specific molecular recognition sites for target analytes, earning them the designation "plastic antibodies" [1]. Despite their significant advantages over biological receptors—including enhanced stability, lower production cost, and reusability—a persistent challenge in MIP technology is non-specific binding. This phenomenon occurs when functional groups located outside the specific imprinted cavities interact indiscriminately with molecules other than the target analyte, thereby reducing selectivity and analytical accuracy [1]. This application note details a strategic approach to mitigate this issue: the electrostatic modification of MIPs using the surfactants SDS (sodium dodecyl sulfate) and CTAB (cetyltrimethylammonium bromide). This methodology effectively blocks non-specific sites, significantly enhancing the selectivity and performance of MIPs in sensing applications [1].
Theoretical Basis of Surfactant Modification
The selective recognition of a target molecule by a MIP is a function of its complementary imprinted cavities and their associated functional groups. However, functional monomers incorporated outside these cavities create non-specific sites that can bind non-target molecules, leading to interference and false-positive signals [1]. Surfactants, being amphiphilic molecules, can electrostatically interact with these exposed functional groups on the MIP surface.
The strategic use of ionic surfactants like SDS (anionic) and CTAB (cationic) capitalizes on this principle. They are designed to interact with and neutralize the charge of functional groups located outside the specific binding cavities. For instance, a MIP based on a cationic polymer like poly(4-vinylpyridine) can be effectively "capped" by the anionic surfactant SDS. Conversely, a MIP based on an anionic polymer like polymethacrylic acid (PMAA) can be blocked using the cationic surfactant CTAB [1]. This interaction forms a surfactant layer that sterically and electrostatically blocks non-specific sites, without occupying the specific, template-shaped cavities, thereby preserving the MIP's intended affinity for its target molecule [1] [26]. Studies have shown that this modification can virtually eliminate non-specific adsorption, a level of performance difficult to achieve by simply increasing template concentration during polymerization [1].
Application Protocols
The following sections provide detailed methodologies for implementing surfactant modification, based on proven experimental work.
Protocol 1: SDS Modification of a Poly(4-Vinylpyridine) MIP
This protocol is designed for MIPs synthesized with 4-vinylpyridine (4VP) as the functional monomer, which yields a polymer with cationic characteristics [1].
- Objective: To eliminate non-specific adsorption on a cationic MIP via electrostatic modification with SDS.
Materials:
- Synthesized MIP and Non-Imprinted Polymer (NIP) particles.
- Sodium Dodecyl Sulfate (SDS), ≥ 85% purity.
- Appropriate buffer or solvent (e.g., deionized water, acetonitrile/water mixtures).
- Laboratory glassware, vortex mixer, and centrifuge.
Procedure:
- Preparation: Weigh out a precise amount (e.g., 40 mg) of the MIP or NIP.
- Surfactant Solution Preparation: Prepare an aqueous SDS solution at a concentration of 6.92 mmol L⁻¹. This concentration is below the critical micellar concentration (CMC) to ensure the presence of surfactant monomers for effective binding [26].
- Modification Incubation: Add 1.00 mL of the SDS solution to the polymer. Incubate the mixture overnight at room temperature under continuous agitation (e.g., on a rocking table).
- Washing: After incubation, filter the polymer suspension through a 0.22 μm nylon membrane.
- Equilibration: Wash the modified polymer with the buffer or solvent to be used in the subsequent binding assay to remove any unbound surfactant.
- The SDS-modified MIP (denoted MIP+-SDS) is now ready for use in binding studies or sensing applications [1] [26].
Protocol 2: CTAB Modification of a Polymethacrylic Acid (PMAA) MIP
This protocol is suitable for MIPs synthesized using methacrylic acid (MAA) as the functional monomer, which produces a polymer with anionic surface properties [1].
- Objective: To eliminate non-specific adsorption on an anionic MIP via electrostatic modification with CTAB.
Materials:
- Synthesized MIP and NIP particles.
- Cetyltrimethylammonium Bromide (CTAB), ≥ 99% purity.
- Appropriate buffer or solvent.
- Laboratory glassware, vortex mixer, and centrifuge.
Procedure:
- Preparation: Weigh out a precise amount (e.g., 40 mg) of the MIP or NIP.
- Surfactant Solution Preparation: Prepare an aqueous CTAB solution at a concentration of 5.49 mmol L⁻¹ (below its CMC) [26].
- Modification Incubation: Add 1.00 mL of the CTAB solution to the polymer and incubate overnight at room temperature with continuous agitation.
- Washing: Filter the polymer suspension through a 0.22 μm membrane.
- Equilibration: Rinse the modified polymer (denoted MIP--CTAB) with the assay buffer to remove excess, unbound CTAB [1] [26].
- The CTAB-modified MIP is now ready for analytical use.
Workflow Visualization
The following diagram illustrates the logical sequence of the electrostatic modification process for both MIP types.
Performance Data and Analysis
The efficacy of surfactant modification is quantitatively demonstrated through binding studies. The tables below summarize key experimental findings.
Table 1: Impact of Surfactant Modification on Binding Affinity (K_eq) for 2,4,5-T MIP [26]
| Solvent System (ACN:H₂O) | No Surfactant (L mol⁻¹) | With SDS (L mol⁻¹) | With CTAB (L mol⁻¹) | With Tween 20 (L mol⁻¹) |
|---|---|---|---|---|
| Pure Acetonitrile | 7.9 × 10⁴ | 9.0 × 10³ | 2.1 × 10⁴ | 7.0 × 10⁴ |
| 40:60 (v/v), φwater=0.83 | 8.3 × 10⁵ | 1.3 × 10⁵ | 2.3 × 10⁴ | 7.5 × 10⁵ |
Table 2: Analytical Performance of Surfactant-Modified MIP for Sulfamethoxazole (SMX) Detection [1]
| Parameter | Unmodified MIP | MIP+-SDS |
|---|---|---|
| Non-Specific Adsorption | Significant | Effectively eliminated |
| Limit of Detection (LOD) | Not Specified | 6 ng mL⁻¹ |
| Stability | -- | Stable at high temperatures |
Key Findings
- Reduction in Non-Specific Binding: Surfactant modification dramatically reduces the binding affinity of both MIPs and NIPs. The reduction is most pronounced with ionic surfactants (SDS and CTAB) compared to non-ionic surfactants like Tween 20, underscoring the dominance of electrostatic mechanisms over hydrophobic ones in this process [26].
- Preservation of Imprinting Effect: While absolute binding affinity decreases, the modification strategy selectively suppresses non-specific binding more effectively than specific binding. This often results in an increased imprinting factor (IF), which is the ratio of the binding affinity of the MIP to the NIP (IF = Keq(MIP)/Keq(NIP)), thereby enhancing the effective selectivity of the polymer [26].
- Analytical Performance: The application of this strategy to a SMX-imprinted MIP resulted in a low detection limit of 6 ng mL⁻¹, demonstrating its utility in developing highly sensitive sensors. The modified MIPs also exhibited excellent stability, making them suitable for on-site applications [1].
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Reagents for Surfactant Modification of MIPs
| Reagent | Function and Rationale |
|---|---|
| Sodium Dodecyl Sulfate (SDS) | Anionic surfactant used to block non-specific sites on cationic MIPs (e.g., poly(4-vinylpyridine)) via electrostatic interaction [1]. |
| Cetyltrimethylammonium Bromide (CTAB) | Cationic surfactant used to block non-specific sites on anionic MIPs (e.g., polymethacrylic acid) via electrostatic interaction [1]. |
| Methacrylic Acid (MAA) | A common functional monomer used in MIP synthesis; forms anionic polymers requiring CTAB for post-modification [1] [27]. |
| 4-Vinylpyridine (4VP) | A common functional monomer used in MIP synthesis; forms cationic polymers requiring SDS for post-modification [1]. |
| Molecularly Imprinted Polymer (MIP) | The core material containing specific recognition cavities for the target analyte and non-specific sites to be blocked. |
| Non-Imprinted Polymer (NIP) | A critical control material, synthesized without the template, used to quantify the extent of non-specific binding [1] [26]. |
Electrostatic modification with SDS and CTAB presents a robust, straightforward, and highly effective strategy for overcoming one of the most significant limitations of MIP technology: non-specific binding. The protocols outlined herein provide researchers with a clear methodology to selectively block interfering sites based on the surface charge of their polymer. This approach significantly enhances the selectivity and analytical performance of MIPs, as evidenced by the quantitative data, facilitating their successful application in complex matrices such as environmental, food, and clinical samples for sensitive and reliable detection. Integrating this surfactant modification step into MIP development protocols is a recommended best practice for advancing biosensing research and applications.
The rational design of molecularly imprinted polymers (MIPs) hinges on the precise selection of functional monomers and solvents, a process critical for creating high-affinity binding sites while minimizing non-specific adsorption. Traditional trial-and-error methods are time-consuming and often yield suboptimal materials. This Application Note details integrated protocols leveraging computational modeling and machine learning (ML) to rationally guide these choices, directly supporting the development of MIPs with enhanced specificity and reduced non-specific binding for applications in biosensing, drug development, and separations.
Computational Modeling for Monomer Selection
Computational approaches allow researchers to predict the strength and nature of interactions between a target molecule (template) and potential functional monomers in silico, before any laboratory synthesis.
Key Methodologies and Workflow
A robust computational strategy combines multiple techniques to evaluate monomer-template compatibility [28] [29].
Table 1: Computational Methods for Monomer Selection
| Method | Primary Function | Key Outputs | Interpretation |
|---|---|---|---|
| Molecular Docking | Identifies optimal binding pose and favorable interaction sites on the template molecule [28] [29]. | Binding affinity (kcal/mol), binding site location. | More negative binding energy indicates stronger interaction. |
| Molecular Dynamics (MD) Simulations | Assesses the stability of the monomer-template complex under simulated conditions [29]. | RMSD, RMSF, Radius of Gyration (Rg), number of hydrogen bonds. | Low RMSD/RMSF and stable H-bonds indicate a robust complex. |
| MM-PBSA/GBSA | Calculates the binding free energy from MD trajectories [29]. | Binding free energy (ΔG bind, kcal/mol). | More negative ΔG bind signifies a more stable and favorable complex. |
| Quantum Chemical Calculations (QCC) | Models electronic structure properties to understand interaction mechanisms [28] [30]. | Interaction energy, electrostatic potential maps. | Higher interaction energy and complementary electrostatic surfaces suggest better monomer choice. |
The following workflow outlines the standard protocol for a computational monomer screening study.
Detailed Experimental Protocol: Computational Monomer Screening
Protocol 1: In Silico Screening of Functional Monomers for a Protein Template (e.g., DJ-1) [29]
- Objective: To identify the most suitable functional monomer for synthesizing a MIP targeting the DJ-1 protein.
- Software Requirements: Molecular modeling software (e.g., AutoDock Vina, GROMACS, Gaussian), visualization tool (e.g., PyMOL, UCSF Chimera).
| Step | Procedure | Parameters & Notes |
|---|---|---|
| 1. Template Preparation | Obtain the 3D structure of the target protein (e.g., PDB ID: 1P5F for DJ-1). Remove water molecules and co-crystallized ligands. Add polar hydrogen atoms and assign partial charges (e.g., using Gasteiger charges). | Ensure the protein structure is complete and protonation states of residues are correct for the intended pH. |
| 2. Monomer Preparation | Draw structures of candidate monomers (e.g., PEDOT, PPy, POAP). Geometry optimization is performed using quantum chemical methods (e.g., Density Functional Theory (DFT) with B3LYP/6-31G* basis set). | For polymers, use oligomer models (e.g., pentamers) to better represent the polymeric state [29]. |
| 3. Molecular Docking | Perform "blind docking" of each monomer against the entire protein surface using AutoDock 4.2 or similar. Use a large grid box to encompass the entire protein. | Run multiple docking simulations (e.g., 100 runs per monomer). Analyze clusters of results to identify the most probable binding pose and its binding energy. |
| 4. Molecular Dynamics (MD) | Solvate the best docked complex in a water box (e.g., SPC water model). Add ions to neutralize the system. Run MD simulation for a sufficient time (e.g., 50-100 ns) using software like GROMACS. | Common force fields: AMBER, CHARMM. Monitor temperature (300 K) and pressure (1 bar) using coupling algorithms. |
| 5. Trajectory Analysis | Calculate Root Mean Square Deviation (RMSD), Root Mean Square Fluctuation (RMSF), Radius of Gyration (Rg), and the number of hydrogen bonds over the simulation trajectory. | A stable RMSD and low RMSF at the binding site indicate a stable complex. Consistent H-bonds suggest specific interactions. |
| 6. Binding Energy Calculation | Use the MM-PBSA (Molecular Mechanics Poisson-Boltzmann Surface Area) method on a set of stable trajectory frames (e.g., last 10 ns) to calculate the binding free energy. | The formula: ΔGbind = Gcomplex - (Gprotein + Gligand). A more negative ΔG_bind indicates a stronger interaction [29]. |
| 7. Decision | Rank monomers based on a combination of docking score, MD stability, and MM-PBSA binding free energy. | For DJ-1, PEDOT was computationally predicted and experimentally verified as the superior monomer [29]. |
Machine Learning for Solvent Optimization
Machine learning models can efficiently navigate the vast chemical space of potential solvents, predicting those that maximize imprinting efficiency and minimize non-specific interactions.
Key ML Approaches and Workflow
ML models learn from existing data to map molecular features of solvents and templates to desired outcomes, such as successful cocrystal formation or high partition coefficients.
Table 2: Machine Learning Approaches for Solvent Screening
| Approach | Application | Key Features | Performance |
|---|---|---|---|
| Interpretable ML (e.g., XGBoost) | Predicting multi-component crystal formation with specific solvents [31]. | Solvent properties (e.g., solubility parameters), API/coformer descriptors, supramolecular synthon motifs. | Accuracy >0.75 for predicting suitable coformers/solvents for unseen molecules [31]. |
| Bayesian Experimental Design | Selecting optimal green solvent mixtures for extracting biomolecules from plant biomass [32]. | Molecular descriptors of solvent mixtures, prior experimental results. | Identifies high-performing solvent candidates with 10-100x fewer experiments. |
| DFT-Pre-trained ML Models | Predicting solvent performance for liquid-liquid extraction (e.g., of 2,3-butanediol) [33]. | Partition coefficients from DFT calculations, molecular structure. | Successfully screened 6717 solvents, with high accuracy validated on a small experimental set (24 solvents). |
The ML-guided solvent screening process is an iterative cycle between prediction and experimental validation.
Detailed Experimental Protocol: ML-Guided Solvent Screening
Protocol 2: Machine Learning Workflow for Solvent Selection in MIP Synthesis [31]
- Objective: To efficiently identify optimal solvents for MIP synthesis that promote specific template-monomer interactions and reduce non-specific binding.
- Requirements: Programming environment (e.g., Python with scikit-learn, XGBoost), chemical featurization libraries (e.g., RDKit), access to automated liquid handling or crystallization workstations for validation.
| Step | Procedure | Parameters & Notes |
|---|---|---|
| 1. Data Set Curation | Compile a high-quality dataset of solvent performance. Use internal data from automated crystallization workstations for consistency. Augment with literature data if available and reliable. | For MIPs, data could include metrics like binding affinity, imprinting factor, and non-specific adsorption for different solvent systems. |
| 2. Feature Calculation | Calculate molecular descriptors for all solvents and template molecules in the dataset. Key features include solubility parameters, molar volume, dipole moment, polarizability, and hydrogen bonding parameters [31]. | Use tools like RDKit or COSMO-RS to generate a comprehensive feature set. Incorporate features related to the template's and monomer's solubility in the solvent. |
| 3. Model Training & Pre-training | Train a model (e.g., XGBoost) on the curated dataset. If in-house data is limited, pre-train the model on a large, general literature dataset, then fine-tune it on the specific in-house data. | The pre-training/fine-tuning framework is effective even with limited proprietary data [31]. Use cross-validation to avoid overfitting. |
| 4. Solvent Prediction & Prioritization | Use the trained model to predict the performance of a vast virtual library of solvents. Rank the solvents based on the predicted outcome (e.g., probability of successful imprinting). | The model can screen thousands of candidates in silico [33]. Generate a shortlist of 20-50 top candidates for experimental testing. |
| 5. Experimental Validation | Test the top-ranked solvents experimentally. For MIPs, this involves synthesizing and characterizing the polymer in each solvent and evaluating key performance metrics (binding capacity, imprinting factor). | Use high-throughput methods to test many conditions rapidly. This step generates new, high-quality data for model refinement. |
| 6. Model Refinement | Feed the experimental results back into the model as new training data. This iterative process continuously improves the model's accuracy and predictive power for future projects. | This active learning loop minimizes the total number of experiments required. |
Integrated Application: Suppressing Non-Specific Binding
A key application of rational selection is designing MIPs with minimal non-specific adsorption. This can be achieved by selecting monomers that form strong, specific complexes with the template and by using surfactants to block non-specific sites.
Table 3: Research Reagent Solutions for MIPs with Reduced Non-Specific Binding
| Reagent | Function/Role | Application Example | Key Finding |
|---|---|---|---|
| Poly(3,4-ethylenedioxythiophene) (PEDOT) | Functional monomer forming strong van der Waals, H-bond, and electrostatic interactions with protein templates [29]. | DJ-1 protein MIP for Parkinson's disease biomarker detection. | Computationally selected PEDOT formed a robust complex with DJ-1, leading to a highly selective MIP sensor [29]. |
| Sodium Dodecyl Sulfate (SDS) | Anionic surfactant used to electrostatically block non-specific binding sites on positively charged MIP surfaces [1]. | Sulfamethoxazole (SMX) MIP for detection in milk and water. | Modification with SDS effectively eliminated non-specific adsorption, achieving a low limit of detection (6 ng mL⁻¹) [1]. |
| Cetyl Trimethyl Ammonium Bromide (CTAB) | Cationic surfactant used to electrostatically block non-specific binding sites on negatively charged MIP surfaces [1]. | Sulfamethoxazole (SMX) MIP. | CTAB modification also eliminated non-specific adsorption, enhancing the selectivity of the MIP [1]. |
| Cetyltrimethylammonium bromide (CTAB) | Pore-regulating agent and surface modifier to improve imprinting efficiency and regenerate sensor surfaces [34]. | Lactate-specific MIP on laser-induced graphene. | Incorporated CTAB to regenerate the sensor surface, improving signal stability and minimizing non-specific binding [34]. |
Protocol 3: Combined Computational and Surfactant-Based Protocol for Low-Background MIPs [29] [1]
- Monomer Selection: Follow Protocol 1 to computationally identify the monomer that forms the most stable and specific pre-polymerization complex with your target analyte (e.g., PEDOT for DJ-1).
- Solvent Screening: Use Protocol 2 to identify a solvent that optimally solvates the template and monomer without disrupting their specific interactions.
- MIP Synthesis & Template Removal: Synthesize the MIP using the selected monomer and solvent. Subsequently, remove the template molecules completely to create specific cavities.
- Surfactant Modification: To suppress residual non-specific binding:
- For a MIP with a net positive surface charge, incubate with the anionic surfactant SDS (e.g., 10 mM solution).
- For a MIP with a net negative surface charge, incubate with the cationic surfactant CTAB (e.g., 10 mM solution).
- Incubation time is typically 30-60 minutes with gentle agitation, followed by rinsing to remove unbound surfactant [1].
- Validation: Characterize the surfactant-modified MIP. Binding isotherms should show high capacity for the target and negligible non-specific adsorption, confirmed by testing against structural analogs.
The pursuit of synthetic receptors with minimized non-specific binding is a central challenge in the development of robust molecularly imprinted polymers (MIPs). Traditional bulk polymerization methods often yield heterogeneous binding sites and require extensive processing, which can introduce variability and limit reproducibility. This application note details two advanced, automated polymerization formats—solid-phase synthesis and automated reactor platforms—designed to overcome these limitations. By providing precise control over reaction parameters and integrating purification into the synthesis workflow, these protocols enable the production of MIPs with enhanced binding site uniformity, significantly reducing non-specific interactions and improving the reliability of subsequent analytical applications.
Solid-Phase Synthesis of MIP Nanoparticles
Principle and Workflow
Solid-phase synthesis employs template molecules immobilized on a solid support. The polymerization occurs directly on this surface, creating binding sites with high accessibility and uniformity. A key advantage is the inherent integration of affinity purification; only high-affinity polymers remain attached to the template and are later eluted, directly addressing the issue of non-specific binding from low-affinity sites or non-imprinted polymers [35]. The entire process, from synthesis to purification, can be completed automatically.
The workflow for the automated solid-phase synthesis of MIP nanoparticles is as follows:
Detailed Experimental Protocol
Objective: To automatically synthesize and purify high-affinity MIP nanoparticles against a target protein (e.g., trypsin, pepsin A, α-amylase) with reduced non-specific binding.
Materials and Equipment:
- Template-Protein Immobilized Glass Beads: Prepared according to established coupling techniques (e.g., 1.7-2.9 nmol of template per gram of glass beads) [35].
- Monomer Mixture: 6.5 mM in water. Common monomers include methacrylic acid (MAA) or acrylamide.
- Initiator System: Ammonium persulfate (APS) and ( N,N,N',N' )-Tetramethylethylenediamine (TEMED).
- Solvents: Deionized water, and elution buffer.
- Automated Reactor: Consisting of a temperature-controlled reactor chamber, a shaking mechanism, a set of pumps for reagent delivery, a fraction collector, and a ( N_2 ) inlet [35].
Procedure:
- Reactor Loading: Load the template-derivatized glass beads (e.g., 20-80 g) into the temperature-controlled reactor chamber.
- Monomer Injection: Use the pump system to deliver the deoxygenated monomer mixture onto the solid phase in the reactor.
- Polymerization Initiation: Inject the initiator solution (APS followed by TEMED) to start the polymerization.
- Polymerization: Maintain the reactor at 15°C with continuous shaking for the desired reaction time (e.g., 2-4 hours is sufficient for high-affinity product) [35].
- Low-Affinity Polymer Removal: Keep the temperature at 15°C and wash the reactor contents. This step removes unreacted monomers, oligomers, and low-affinity MIP nanoparticles, which are diverted to waste.
- High-Affinity MIP Elution: Increase the reactor temperature to 60°C and wash with elution buffer. The elevated temperature disrupts the interactions between the high-affinity MIP nanoparticles and the immobilized template, releasing them into the eluent.
- Product Collection: The fraction collector is used to gather the eluate containing the purified, high-affinity MIP nanoparticles.
- Characterization: Analyze the collected MIP nanoparticles for size (e.g., DLS, TEM), concentration, and binding affinity (e.g., SPR) [35].
Typical Outcomes: The table below summarizes quantitative data from a typical automated solid-phase synthesis for different protein templates [35].
Table 1: Performance Data for Automatically Synthesized MIP NPs
| Target Protein | Hydrodynamic Diameter (nm) | Dissociation Constant (K_D) | Process Yield (%, w/w) |
|---|---|---|---|
| Pepsin A | 208 ± 3 | 1.7 × 10⁻¹¹ M | 8 - 10.5 (with 20g solid phase) |
| Trypsin | 207 ± 12 | 4.1 × 10⁻¹¹ M | 8 - 10.5 (with 20g solid phase) |
| α-Amylase | 236 ± 4 | 3.4 × 10⁻¹⁰ M | 8 - 10.5 (with 20g solid phase) |
Automated Synthesis via the Chemputer Platform
Principle and Workflow
The Chemputer is a universal, programmable chemical synthesizer that automates multi-step liquid handling and reaction sequences using the Chemical Description Language (χDL) [36]. Its modular architecture allows for the automation of complex processes, including solid-phase peptide synthesis (SPPS) and subsequent chemical modifications, with exceptional reproducibility. This platform is highly adaptable for MIP research, enabling the precise and automated exploration of monomer formulations, polymerization conditions, and workup procedures, which is critical for systematically optimizing MIP performance and minimizing batch-to-batch variability.
The automated workflow on the Chemputer platform is as follows:
Detailed Experimental Protocol for Automated Synthesis
Objective: To execute a fully automated, multi-step synthesis—such as a peptide-based MIP precursor—with in-line purification, demonstrating high reproducibility and the integration of specific modifications.
Materials and Equipment:
- Chemputer Platform: Configured with modules including an SPPS reactor (fritted filter), a precipitating unit, reagent bottles, solvent pumps, and a pneumatic controller for nitrogen/vacuum [36].
- Solvents and Reagents: Anhydrous DMF, diethyl ether, cleavage cocktail (e.g., TFA/H₂O/TIPS, 90:5:5), piperidine solution, Fmoc-amino acids, coupling reagents (e.g., HATU), and base (e.g., DIPEA).
- χDL Script: A digitally encoded procedure defining all operational steps (Add, Purge, Filter, etc.) and parameters (reaction times, temperatures, volumes) [36].
Procedure (Exemplified for Peptide Synthesis):
- System Initialization: The Chemputer executes the χDL script, initializing all modules and pre-chilling the precipitating unit.
- Resin Swelling: The SPPS reactor is charged with resin, which is then swollen with a specified volume of DMF.
- Iterative Elongation Cycle:
- Fmoc Deprotection: The resin is treated with 20% piperidine in DMF (e.g., 2 × 9 mL, 15 min total) and subsequently washed with DMF.
- Amino Acid Coupling: A solution of Fmoc-protected amino acid (e.g., 0.5 M in DMF), coupling reagent HATU (e.g., 0.45 M in DMF), and base DIPEA is added to the reactor. The coupling proceeds for a set time (e.g., 1 × 30 min) with nitrogen bubbling for mixing, followed by washing [36].
- This deprotection/coupling cycle repeats for each amino acid in the sequence.
- Final Cleavage and Deprotection: A cleavage cocktail (e.g., TFA/H₂O/TIPS) is added to the reactor and mixed for a defined period (e.g., 2 hours) to simultaneously cleave the peptide from the resin and remove protecting groups.
- Precipitation and Purification:
- The TFA cleavage mixture is transferred to the pre-chilled precipitating unit containing cold diethyl ether (-20°C).
- The mixture is agitated (e.g., via nitrogen bubbling) for 30 minutes to precipitate the product.
- The ether is filtered away, and the precipitate is washed multiple times with fresh cold ether.
- The final product is dried under vacuum, dissolved in a suitable solvent (e.g., MeCN/H₂O), and transferred to a product flask [36].
- Analysis: The product is analyzed by RP-HPLC and ESI-MS.
Typical Outcomes: Automated synthesis on platforms like the Chemputer demonstrates high reproducibility and efficiency. The table below shows representative data for peptides synthesized under such automated protocols [36].
Table 2: Performance Data for Compounds Synthesized via Automated Platforms
| Synthesized Compound | Crude Purity | Reported Yield | Key Quantitative Metric |
|---|---|---|---|
| ACP(65-74) | >87% | 41% (53 mg) | Benchmark sequence [36] |
| 18A (Amphipathic Peptide) | >87% | 67% (195 mg) | Demonstrates handling of longer sequences [36] |
| GHRH(1-29) | >87% | 59% (261 mg) | Complex peptide in high yield [36] |
| N-Methyl-18A | 79% | 62% | Incorporation of sterically hindered monomers [36] |
The Scientist's Toolkit: Research Reagent Solutions
This table details essential materials and their functions in the described advanced polymerization formats.
Table 3: Key Reagents for Advanced Polymerization Formats
| Reagent / Material | Function / Role | Application Notes |
|---|---|---|
| Template-Immobilized Beads | Solid support providing defined, accessible binding sites during synthesis and in-situ affinity purification. | Critical for solid-phase synthesis; ensures high-affinity product and removes low-affinity polymers [35]. |
| Functional Monomers | Interact with the template to create complementary binding cavities. | Methacrylic acid (MAA) is common for hydrogen bonding; 4-vinylpyridine for acidic targets [37] [1]. |
| Cross-linker | Creates a rigid 3D polymer network to stabilize the imprinted binding sites. | EGDMA is widely used. High cross-linking density ensures stability and reusability [37]. |
| Initiator System | Starts the radical polymerization reaction. | APS/TEMED for aqueous systems; AIBN for organic solvents [35] [27]. |
| Porogenic Solvent | Dictates porosity and influences monomer-template interactions during polymerization. | Solvent polarity (e.g., acetonitrile, DMSO) is tuned to optimize cavity formation and binding kinetics [37]. |
| Surfactants | Post-synthesis modifiers to suppress non-specific binding. | SDS or CTAB can be used to block external functional groups on MIPs, enhancing selectivity [1]. |
| Chemical Description Language (χDL) | Digitally encodes and automates complex synthetic procedures. | Enables reproducibility and precise execution of multi-step protocols on platforms like the Chemputer [36]. |
Molecularly imprinted polymers (MIPs) are synthetic, highly cross-linked polymers with tailor-made recognition sites complementary to a target molecule in shape, size, and functional groups [38]. Their role as robust, stable, and cost-effective artificial antibodies makes them particularly suited for applications in complex biological matrices, where natural receptors often fail due to instability or non-specific binding [39] [38]. This document details specific application notes and experimental protocols, framed within a broader thesis on reducing non-specific binding, to guide researchers in deploying MIP technology for biosensing, bioanalysis, and targeted drug delivery.
Application Notes & Case Studies
The following case studies demonstrate the performance of advanced MIP systems in complex biological and environmental samples.
Table 1: Case Study Summary in Biosensing and Bioanalysis
| Case Study | Target Analyte | Matrix | MIP Format/Sensor Platform | Key Performance Metrics | Reference |
|---|---|---|---|---|---|
| ACS 1: Therapeutic Drug Monitoring | Paracetamol | Human Plasma | Electrochemical Sensor (MIP NP-based) | Selective detection in plasma; high stability | [40] [41] |
| ACS 2: Cancer Biomarker Detection | Prostate-Specific Antigen (PSA) | Biological Samples | MIP-based Biosensor | High selectivity for PSA trapping | [41] |
| ACS 3: Animal Drug Residue Analysis | Olaquindox & its Metabolite | Animal Products | Magnetic MIP NPs + Emulsion Polymerization | Selective & simultaneous detection | [40] |
| ACS 4: Pre-concentration for Analysis | Captopril | Rat Plasma | Cu²⁺ Mediated Magnetic MIP NPs (Sol-Gel) | Selective enrichment of trace amounts | [40] |
| ACS 5: Toxic Gas Adsorption | H₂S (using H₂O as template) | Industrial Gas Streams | Core-Shell MIL-101(Cr)@MIPs | 94.3% adsorption efficiency; superior selectivity over CO₂/CH₄ | [42] |
Table 2: Case Study Summary in Drug Delivery
| Case Study | Active Ingredient | Polymer System | Key Findings | In Vitro/Ex Vivo Model | Reference |
|---|---|---|---|---|---|
| ACD 1: Targeted Cancer Therapy | 5-Fluorouracil (5-FU) | MIP via Free Radical Polymerization | 82% encapsulation efficiency; sustained release at pH 7.4 & 5.5; >80% lysosomal co-localization | MCF-7 & HCT-116 cancer cells | [43] |
| ACD 2: Sustained Drug Delivery | Theophylline | Cross-linked MIP | More sustained release compared to Non-Imprinted Polymer (NIP); high binding affinity | N/A | [39] |
Detailed Experimental Protocols
Protocol 1: Computational Design of High-Affinity MIPs
Rational design is crucial for creating MIPs with high affinity and reduced non-specific binding [19] [44]. This protocol uses molecular modeling for monomer selection.
3.1.1 Procedure
- Template Preparation: Obtain the 3D structure of the target molecule from databases like PubChem or ZINC. If the target is a macromolecule (e.g., protein), select an appropriate epitope. Energy-minimize the structure using a force field (e.g., Tripos force field) [44].
- Automated Monomer Screening: Use molecular modeling software (e.g., SYBYL) to screen a virtual library of commercially available functional monomers. The script should sequentially dock each monomer to the template, calculating the stabilization energy (ΔE) of the complex [44].
- Monomer Selection & Ratio Optimization: Rank monomers based on their ΔE values. Select the top monomers with the strongest interactions. Further, use molecular dynamics (MD) simulations to model the pre-polymerization mixture containing the template, selected monomers, cross-linker, and solvent. Analyze radial distribution functions (RDFs) to optimize the monomer-to-template ratio for stable complex formation [44].
The following workflow outlines the key stages of the computational design process.
Protocol 2: Solid-Phase Synthesis of MIP Nanoparticles (MIP NPs)
Solid-phase synthesis yields uniform MIP NPs with excellent binding affinity and minimal template leakage, ideal for biological applications [39] [38].
3.2.1 Materials
- Template: Target molecule or structural analogue (dummy template).
- Functional Monomer: Selected from computational design (e.g., methacrylic acid).
- Cross-linker: Ethylene glycol dimethacrylate (EGDMA).
- Initiator: 2,2'-Azobisisobutyronitrile (AIBN).
- Solid Support: Glass beads or silica particles.
- Coupling Agent: e.g., (3-Aminopropyl)triethoxysilane (APTES).
- Solvent: Acetonitrile or toluene.
3.2.2 Procedure
- Immobilize Template: Covalently couple the template molecule onto the surface of the solid support beads using an appropriate coupling agent [38].
- Prepare Polymerization Mixture: In a sealed vial, dissolve the functional monomer, cross-linker (EGDMA), and initiator (AIBN) in a suitable solvent. Degas the mixture with nitrogen or argon for 10 minutes to remove oxygen [38].
- Polymerization: Add the polymerization mixture to the template-immobilized beads. Incubate with agitation (e.g., on a roller) at 60°C for 12-24 hours to initiate thermal polymerization [38].
- Template Removal & Washing: After polymerization, transfer the beads to a chromatography column. Wash extensively with a mild acid, organic solvent, or Soxhlet extraction to remove the template, leaving behind specific cavities [39] [38].
- Elute MIP NPs: Recover the MIP NPs from the solid support by gentle physical agitation or by cleaving a labile covalent bond. Finally, wash and characterize the nanoparticles [38].
Protocol 3: Development of a MIP-based Electrochemical Sensor
MIPs serve as robust recognition elements in sensors for detecting analytes in complex fluids like plasma or urine [24] [45] [41].
3.3.1 Sensor Fabrication
- Electrode Preparation: Clean the working electrode (e.g., glassy carbon, gold, or screen-printed carbon electrode) according to standard protocols.
- MIP Immobilization: Deposit the synthesized MIP (e.g., MIP NPs from Protocol 2) onto the electrode surface. This can be achieved by:
- Blocking: Treat the modified electrode with a blocking agent (e.g., bovine serum albumin, BSA) to passivate any remaining non-specific binding sites on the electrode surface, a critical step for reducing non-specific binding [45].
3.3.2 Measurement & Detection
- Rebinding: Incubate the modified electrode in the sample solution containing the target analyte for a predetermined time.
- Electrochemical Measurement: After washing, transfer the electrode to a clean electrochemical cell containing a redox probe (e.g., [Fe(CN)₆]³⁻/⁴⁻). Measure the electrochemical signal (e.g., via differential pulse voltammetry (DPV) or electrochemical impedance spectroscopy (EIS)).
- Quantification: The binding of the target analyte to the MIP layer hinders the access of the redox probe to the electrode surface, resulting in a measurable change in current or impedance. This change is correlated with the analyte concentration [45].
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Reagents for MIP Research and Development
| Reagent Category | Specific Examples | Function/Purpose |
|---|---|---|
| Functional Monomers | Methacrylic acid (MAA), Acrylamide, Vinylpyridine | Interact with template via non-covalent bonds (H-bonding, ionic) to form recognition sites. |
| Cross-linkers | Ethylene glycol dimethacrylate (EGDMA), Trimethylolpropane trimethacrylate (TRIM) | Create a rigid 3D polymer network to stabilize the imprinted cavities. |
| Initiators | 2,2'-Azobisisobutyronitrile (AIBN), Ammonium persulfate (APS) | Generate free radicals to initiate the polymerization reaction (thermally or UV-induced). |
| Solvents/Porogens | Acetonitrile, Chloroform, Toluene, Dimethylformamide (DMF) | Dissolve all polymerization components and create pore structure within the polymer. |
| Template Molecules | Target drug, biomarker, hormone, or a structural analogue (dummy template) | Serves as the "mold" for creating specific binding cavities complementary to the target. |
| Solid Supports | Glass beads, Silica particles | Used in solid-phase synthesis to immobilize the template and produce uniform MIP NPs. |
The case studies and protocols presented herein demonstrate that MIPs, through rational design and advanced synthesis strategies, are capable of functioning with high specificity and efficacy in complex matrices. The integration of computational design, solid-phase synthesis, and sensor technology provides a powerful framework for developing next-generation MIPs with minimized non-specific binding. This advancement is pivotal for their successful translation into real-world clinical, analytical, and therapeutic applications, from point-of-care diagnostics to targeted drug delivery systems.
Optimizing MIP Performance and Overcoming Practical Challenges
Molecularly imprinted polymers (MIPs) are synthetic materials engineered with specific molecular recognition capabilities, functioning as "plastic antibodies" for targeted analyte binding. [1] The systematic optimization of their synthesis parameters—functional monomers, cross-linkers, and solvents—is fundamental to developing high-performance MIPs with enhanced selectivity and minimized non-specific binding, a core challenge in the field. [1] [46] Non-specific adsorption, often stemming from functional groups located outside the imprinted cavities, significantly compromises the performance of MIPs in sensing and separation applications. [1] This protocol details advanced strategies for optimizing MIP synthesis, focusing on creating selective binding sites while actively suppressing non-specific interactions, thereby contributing to the broader thesis goal of developing superior MIPs for diagnostic and therapeutic applications.
Quantitative Optimization Parameters
The tables below consolidate critical quantitative data from recent studies to guide the selection of synthesis parameters.
Table 1: Optimized Functional Monomers and Cross-linkers for Target Analytics
| Target Analyte | Functional Monomer | Cross-linker | Optimal Molar Ratio (Template:Monomer:Cross-linker) | Key Performance Metric | Reference |
|---|---|---|---|---|---|
| Terbium (Tb³⁺) / Lead (Pb²⁺) | Methacrylic acid (MAA), 2-Vinylpyridine (2-VP) | Divinylbenzene (DVB) | 1:5:5:8 (Template:Monomer 1:Monomer 2:Cross-linker) | Imprinting Factor: 7.06 | [47] |
| Paracetamol | Deep Eutectic Solvent (ChCl:MAA) | EGDMA | 1:4:20 | Binding Capacity: 8.90 mg/g; Imprinting Factor: 3.85 | [48] |
| Levofloxacin (LEV) | Methacrylic Acid (MAA) | EGDMA | 1:3:16 (in Ethanol:DMSO) | Removal Efficiency: 99.15%; Imprinting Factor: 3.36 | [27] |
| Emtricitabine | 2-Vinylpyridine (2-VP) | EGDMA | 1: ~10.8: ~104 (by mmol mass) | High selectivity in wastewater; Reusable for 5 cycles | [49] |
| Sulfamethoxazole (SMX) | 4-Vinylpyridine / MAA | EGDMA | Specific ratio not provided | High selectivity post-surfactant modification | [1] |
Table 2: Solvent Systems and Porogens in MIP Synthesis
| Solvent System (Ratio) | Polymerization Method | Target Analyte | Impact on MIP Performance | Reference |
|---|---|---|---|---|
| Toluene | Bulk Polymerization | Terbium/Lead, Emtricitabine | Non-polar porogen promoting porous structure formation | [47] [49] |
| Ethanol : Dimethyl Sulfoxide (DMSO) (40:20 mL) | Precipitation Polymerization | Levofloxacin (LEV) | Superior removal efficiency (99.15%) and high imprinting factor | [27] |
| Ethanol : Acetonitrile (30:30 mL) | Precipitation Polymerization | Levofloxacin (LEV) | Moderate performance for comparison | [27] |
| Acetonitrile | Bulk Polymerization | Paracetamol | Standard solvent for DES-MIP synthesis | [48] |
Experimental Protocols
Protocol 1: Optimization via Computational Screening and Modeling
Rational design using computational tools reduces reliance on trial-and-error and predicts monomer-template affinity. [19] [46]
Molecular Dynamics (MD) Simulations:
- Objective: To simulate and monitor the behavior and self-assembly of functional monomers and cross-linkers around the template molecule in a virtual environment. [46]
- Procedure: Use software (e.g., GROMACS, AMBER) to model the pre-polymerization mixture. Analyze the stability and energy of the forming template-monomer complexes to identify the most promising functional monomers.
Binding Energy Calculations:
- Objective: To quantitatively calculate the binding energy between the target molecule and potential functional monomers. [46]
- Procedure: Employ computational chemistry software (e.g., Gaussian) to perform density functional theory (DFT) calculations. Compare the binding energies of different monomer candidates; a higher (more negative) binding energy typically indicates a stronger and more stable complex.
Experimental Validation:
- Procedure: Synthesize MIPs with the top-ranked monomers from the computational screening. Compare their binding capacity and imprinting factor against MIPs made with poorly ranked monomers to validate the model. Techniques like nuclear magnetic resonance (NMR) spectroscopy or calorimetric titration can further experimentally probe monomer-template interactions. [46]
Protocol 2: Suppression of Non-Specific Adsorption via Surfactant Modification
This protocol details a post-synthesis strategy to chemically mask non-specific sites, significantly enhancing selectivity. [1]
Materials:
- Synthesized MIP particles (e.g., poly(4-vinylpyridine) or polymethacrylic acid-based).
- Surfactants: Sodium dodecyl sulfate (SDS) for modifying MIPs with external positive charges, or Cetyl trimethyl ammonium bromide (CTAB) for modifying MIPs with external negative charges. [1]
- Deionized water.
Procedure:
- Weigh 100 mg of the synthesized MIP.
- Prepare a 1 mM surfactant solution (SDS or CTAB) in deionized water.
- Incubate the MIP particles with 10 mL of the surfactant solution for 30 minutes under gentle stirring. The surfactant molecules will electrostatically bind to the functional groups located on the polymer's external surface, which are responsible for non-specific binding.
- Recover the modified MIP particles (now designated MIP±-SDS/CTAB) by centrifugation or filtration.
- Wash the particles gently with deionized water to remove any unbound surfactant.
- Validate the modification by comparing the binding isotherms of the target molecule (e.g., Sulfamethoxazole) on the modified MIP and its corresponding Non-Imprinted Polymer (NIP). Successful modification is indicated by a significant reduction in the NIP's adsorption capacity while the MIP's capacity remains high. [1]
Protocol 3: Formulation Optimization using Response Surface Methodology (RSM)
RSM is a statistical technique for efficiently optimizing multiple synthesis parameters simultaneously. [48]
Experimental Design:
- Objective: To optimize the concentrations of template (Paracetamol), functional monomer (DES), and cross-linker (EGDMA) for maximum binding capacity.
- Design: Employ a Central Composite Design (CCD) using software like Design-Expert. Define the independent variables and their ranges (e.g., Template: 1-4 mmol, Functional Monomer: 4-8 mmol, Cross-linker: 15-20 mmol). [48]
Polymer Synthesis and Evaluation:
- Synthesize MIPs according to all the experimental runs generated by the CCD model, typically using bulk polymerization. [48]
- For each synthesized MIP, perform a batch binding assay. Incubate a fixed amount of polymer (e.g., 10 mg) with a standard solution of the target analyte (e.g., 10 mL of 10 ppm Paracetamol). [48]
- Shake the mixture for a predetermined time (e.g., 2 h), then filter and analyze the supernatant concentration using a UV-Vis spectrophotometer.
- Calculate the binding capacity (Q, mg/g) for each MIP formulation.
Data Analysis and Model Validation:
- Input the experimental binding capacities into the RSM software. Perform analysis of variance (ANOVA) to assess the model's significance and identify which factors have a statistically significant impact on the binding capacity.
- Use the model to predict the optimal molar ratio for synthesis. Synthesize a new MIP at this predicted optimum and experimentally verify its binding capacity to confirm the model's accuracy. [48]
Workflow Visualization
The following diagram illustrates the logical workflow for the systematic optimization of MIP synthesis.
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Materials for MIP Synthesis and Optimization
| Reagent Category | Specific Examples | Function in MIP Synthesis |
|---|---|---|
| Functional Monomers | Methacrylic Acid (MAA), 2-Vinylpyridine (2-VP), 4-Vinylpyridine, Deep Eutectic Solvents (e.g., ChCl:MAA) | Interact with the template molecule via non-covalent bonds (H-bonding, ionic, van der Waals) to form a pre-polymerization complex. The choice dictates complementarity. |
| Cross-linkers | Ethylene Glycol Dimethacrylate (EGDMA), Divinylbenzene (DVB), Trimethylolpropane Trimethacrylate (TRIM) | Creates a rigid, porous 3D polymer network that stabilizes the binding cavities after template removal. |
| Initiators | Azobisisobutyronitrile (AIBN), 1,1´-Azobis(cyclohexanecarbonitrile), Benzoyl Peroxide (BPO) | Generates free radicals upon thermal or UV decomposition to initiate the polymerization reaction. |
| Porogens/Solvents | Toluene, Acetonitrile, Dimethyl Sulfoxide (DMSO), Ethanol, solvent mixtures | Dissolves all components and creates pores within the polymer matrix, influencing morphology and template accessibility. |
| Suppression Agents | Sodium Dodecyl Sulfate (SDS), Cetyl Trimethyl Ammonium Bromide (CTAB) | Modifies external polymer surface to electrostatically block functional groups responsible for non-specific binding. |
| Characterization Tools | FTIR, SEM, TGA, BET Surface Area Analyzer, UV-Vis Spectrophotometer | Confirms successful synthesis, analyzes morphology, thermal stability, porosity, and quantifies binding performance. |
Balancing Selectivity and Catalytic Activity in Hybrid Nanozyme@MIP Systems
The integration of molecularly imprinted polymers (MIPs) with nanozymes represents a cutting-edge approach in biomimetic sensing, aiming to synergize the superior selectivity of MIPs with the catalytic prowess of nanozymes. MIPs are synthetic polymers designed with specific cavities that complement the target molecule in shape, size, and chemical functionality, earning them the moniker "plastic antibodies" [1]. Nanozymes are nanomaterial-based artificial enzymes that mimic the catalytic functions of natural enzymes but offer advantages in cost, stability, and mass production [50]. A critical challenge in this hybrid system is the persistent issue of non-specific adsorption in MIPs, where functional groups outside the imprinted cavities bind non-target molecules, thereby compromising selectivity and analytical accuracy [1] [16]. This Application Note outlines validated protocols to suppress non-specific binding while preserving high catalytic activity, providing a reliable framework for researchers developing advanced biosensors.
Experimental Protocols
Synthesis of Molecularly Imprinted Polymers (MIPs)
Objective: To synthesize MIPs with specific recognition sites for a target analyte while establishing a protocol that minimizes non-specific binding from the outset.
Materials:
- Template Molecule: Target analyte (e.g., Sulfamethoxazole (SMX) [1] or a microcystin variant [51]).
- Functional Monomers: Methacrylic acid (MAA) or 4-vinylpyridine [1].
- Cross-linker: Ethylene glycol dimethacrylate (EGDMA) [1].
- Initiator: Ammonium persulfate (APS) [1].
- Solvent: Appropriate porogenic solvent (e.g., ethanol, dimethylsulfoxide (DMSO)) [1].
Procedure:
- Pre-complexation: Dissolve the template molecule and functional monomer in the porogenic solvent. Allow the mixture to incubate to facilitate complex formation via non-covalent interactions [1].
- Polymerization: Add the cross-linker and initiator to the pre-complexed solution. Purge the reaction mixture with an inert gas (e.g., nitrogen) to remove oxygen.
- Initiation: Initiate polymerization using thermal or UV radiation methods, depending on the initiator used [1].
- Template Removal: After polymerization, wash the polymer extensively with a suitable solvent (e.g., methanol-acetic acid mixture) to remove the template molecule, thereby creating specific recognition cavities [1].
- Drying: Dry the resulting MIP particles under vacuum for further use or modification.
Electrostatic Suppression of Non-Specific Adsorption
Objective: To eliminate non-specific adsorption by blocking functional groups outside the imprinted cavities using surfactant modification.
Rationale: Non-specific adsorption is primarily caused by functional monomers located outside the specific cavities. Surfactants can electrostatically interact with and block these external groups without disrupting the internal imprinted sites [1] [16].
Materials:
- Synthesized MIPs (e.g., poly(4-vinylpyridine) or polymethacrylic acid-based) [1].
- Surfactants: Sodium dodecyl sulfate (SDS) or Cetyl trimethyl ammonium bromide (CTAB) [1].
- Solvent: Deionized water or buffer.
Procedure:
- Surfactant Selection: Choose a surfactant with a charge opposite to that of the external functional groups of the MIP.
- Modification: Incubate the MIPs with a solution of the selected surfactant (e.g., 1-5 mM) for a predetermined time (e.g., 30-60 minutes) with gentle agitation [1].
- Washing: Centrifuge and wash the modified MIPs (MIP-SDS or MIP-CTAB) with the solvent to remove any unbound surfactant.
- Validation: The success of modification is confirmed by a significant reduction in the binding capacity of the corresponding Non-Imprinted Polymer (NIP), which lacks specific cavities and primarily exhibits non-specific binding [1].
Fabrication of Hybrid Nanozyme@MIP Systems
Objective: To integrate a catalytically active nanozyme with the selective MIP to create a hybrid sensor material.
Materials:
- Surfactant-modified MIPs.
- Nanozyme material (e.g., Ferromagnetic nanoparticles (Fe₃O₄), Au nanoparticles, CeO₂ nanoparticles, or Metal-Organic Frameworks (MOFs) with peroxidase-like or oxidase-like activity) [50].
- Coupling agents if required.
Procedure: Two primary strategies can be employed:
- Surface Imprinting on Nanozymes: Directly form the MIP layer on the surface of the nanozyme. This method ensures the catalytic core is intimately connected to the selective shell [50].
- The nanozyme is treated as the solid support during the MIP synthesis protocol (Section 2.1).
- This creates a core-shell structure where the nanozyme is the core, and the imprinted polymer is the shell.
- Post-synthesis Integration: Physically mix or chemically conjugate the pre-synthesized, surfactant-modified MIPs with the nanozyme material. This approach offers flexibility but may result in a less homogeneous structure.
The following diagram illustrates the fabrication workflow and the mechanism of the resulting hybrid material.
Diagram Title: Hybrid Nanozyme@MIP Fabrication and Function
Characterization and Performance Data
Quantitative Assessment of Selectivity and Catalytic Activity
The performance of the hybrid system should be rigorously tested. The table below summarizes key metrics from a model system targeting Sulfamethoxazole (SMX), demonstrating the effect of surfactant modification.
Table 1: Performance Metrics of Surfactant-Modified MIPs for Sulfamethoxazole (SMX) Detection
| Material | Adsorption Capacity (SMX) | Non-Specific Adsorption | Limit of Detection (LOD) | Key Finding |
|---|---|---|---|---|
| MIP | Higher | Present | Not Specified | Confirms cavity-specific binding [1] |
| Non-Imprinted Polymer (NIP) | Lower | Significant (Baseline) | Not Applicable | Measures non-specific background [1] |
| MIP-SDS | Retained High | Effectively Eliminated | 6 ng mL⁻¹ | High selectivity and sensitivity achieved [1] [16] |
| MIP-CTAB | Retained High | Effectively Eliminated | Data in source | High selectivity confirmed [1] |
Table 2: Catalytic Activities of Common Nanozymes for Signal Generation
| Nanozyme Material | Enzyme-like Activity | Typical Application in Sensing | Reference |
|---|---|---|---|
| Fe₃O₄ Nanoparticles | Peroxidase | Oxidizes chromogenic substrates (e.g., ABTS, TMB) in presence of H₂O₂ [50] | [50] |
| Au Nanoparticles | Oxidase, Peroxidase | Oxidizes substrates like TMB without H₂O₂ (oxidase) or with H₂O₂ (peroxidase) [50] | [50] |
| CeO₂ Nanoparticles | Oxidase, Superoxide Dismutase | Oxidizes substrates using molecular oxygen [50] | [50] |
| Metal-Organic Frameworks (MOFs) | Peroxidase, Laccase | High surface area and tunable porosity enhance catalytic efficiency [50] [52] | [50] [52] |
Validation in Complex Matrices
The practical utility of the MIP-SDS system was demonstrated by detecting SMX in milk and water samples, achieving promising recovery rates, which confirms the method's robustness against matrix effects in real-world samples [1]. Furthermore, the modified MIPs remained stable at high temperatures, underscoring their suitability for on-site applications [1].
The Scientist's Toolkit: Essential Research Reagents
This section provides a curated list of critical reagents for developing hybrid Nanozyme@MIP systems, based on the protocols and studies cited.
Table 3: Key Research Reagent Solutions for Nanozyme@MIP Development
| Reagent / Material | Function / Role | Specific Example & Rationale |
|---|---|---|
| Functional Monomers | Forms interactions with the template; defines chemical complementarity of cavities. | Methacrylic acid (MAA), 4-Vinylpyridine. Choice depends on template chemistry (H-bond donor/acceptor) [1]. |
| Cross-linker | Creates rigid polymer network; stabilizes imprinted cavities. | Ethylene glycol dimethacrylate (EGDMA). Provides mechanical stability and defines porosity [1]. |
| Surfactants | Suppresses non-specific adsorption by blocking external functional groups. | SDS (anionic), CTAB (cationic). Selected based on charge complementarity with MIP surface [1] [16]. |
| Nanozymes | Provides catalytic signal amplification for detection. | Fe₃O₄ NPs (peroxidase-mimic), Au NPs (oxidase-mimic). Chosen for high activity and stability in sensor setups [50]. |
| Chromogenic Substrates | Visual or spectroscopic signal output for catalytic activity. | ABTS, TMB. Produce a colored product upon oxidation by peroxidase/oxidase-like nanozymes [50]. |
Application Workflow in Biosensing
A typical workflow for using a hybrid Nanozyme@MIP in a biosensor for a target analyte (e.g., a toxin, biomarker, or drug) involves the following stages, which leverage the material's dual functions:
Diagram Title: Biosensing Workflow with Hybrid Material
- Selective Binding: The sample containing the target analyte is introduced to the hybrid Nanozyme@MIP material. The MIP shell selectively captures and concentrates the target molecule within its cavities.
- Catalytic Signal Generation: The presence of the target can modulate the activity of the nanozyme core, or the system is simply used as a selective capture probe. A signal-generating substrate (e.g., TMB for a peroxidase-mimic) is added. The nanozyme core catalyzes a reaction, producing a measurable signal (e.g., color change, fluorescence, electrical current).
- Signal Detection: The generated signal is quantified using appropriate instrumentation (e.g., spectrophotometer, smartphone camera, potentiostat), providing a concentration-dependent measurement of the target analyte.
This integrated workflow ensures that the detected signal originates specifically from the target-bound hybrid material, minimizing false positives from non-specific adsorption.
Addressing Template Leaching and Binding Site Heterogeneity
Molecularly imprinted polymers (MIPs) are synthetic biomimetic receptors with tailor-made binding sites complementary to target molecules in shape, size, and functional groups [53]. Despite their significant advantages over biological recognition elements, two persistent challenges hinder their reliable application: template leaching and binding site heterogeneity. Template leaching refers to the incomplete removal of the original template molecule during MIP preparation, which subsequently releases during analytical applications, causing false positives and accuracy issues [54]. Binding site heterogeneity describes the presence of binding sites with varying affinities and selectivities within the MIP matrix, leading to nonlinear binding behavior and reduced specificity [55]. This application note details protocols and strategies to address these critical challenges, enabling researchers to produce more reliable MIPs for diagnostic and pharmaceutical applications.
Experimental Protocols
Protocol 1: Surfactant Modification to Suppress Non-Specific Binding
Principle: Electrostatic modification of MIPs with surfactants masks external functional groups responsible for non-specific adsorption while preserving the recognition properties of imprinted cavities [1].
Materials:
- Synthesized MIP particles (e.g., poly(4-vinylpyridine) or polymethacrylic acid-based)
- Sodium dodecyl sulfate (SDS)
- Cetyl trimethyl ammonium bromide (CTAB)
- Deionized water
- Methanol or ethanol
- Ultrasonic bath
- Vacuum filtration setup
Procedure:
- Preparation of Surfactant Solutions: Prepare 10 mM aqueous solutions of SDS and CTAB separately.
- MIP Modification: Disperse 100 mg of finely crushed MIP particles in 20 mL of the surfactant solution (Use SDS for modifying positively charged MIPs like poly(4-vinylpyridine) and CTAB for negatively charged MIPs like polymethacrylic acid).
- Incubation: Place the mixture in an ultrasonic bath for 15 minutes, then stir magnetically for 2 hours at room temperature.
- Washing: Recover the modified MIP particles (denoted as MIP+-SDS or MIP--CTAB) by vacuum filtration.
- Rinsing: Wash the particles three times with 10 mL of a 50:50 (v/v) water-methanol solution to remove physically adsorbed surfactant.
- Drying: Dry the modified MIPs under vacuum at 40°C overnight before use or storage.
Key Considerations: This treatment has been shown to effectively eliminate non-specific adsorption while maintaining the specific binding capacity of the imprinted cavities, resulting in improved selectivity and a lower limit of detection [1].
Protocol 2: Optimized Chemical Extraction for Template Removal
Principle: Sequential washing with solvents of varying polarity and acidity disrupts the interactions between the template and the functional monomers, facilitating complete template removal while preserving cavity integrity [56] [54].
Materials:
- MIP in monolithic or finely crushed form
- Methanol
- Acetic acid
- Deionized water
- Soxhlet extraction apparatus (optional)
- Orbital shaker or ultrasonic bath
Procedure:
- Initial Wash: Place the MIP in a container and incubate with a mild solvent (e.g., methanol or acetone) for 1 hour with stirring to remove the bulk of the template and unreacted components.
- Acidic Solvent Extraction: Subject the MIP to a more stringent washing solution, typically methanol:acetic acid (9:1, v/v). This can be done via:
- Soxhlet Extraction: Transfer the MIP to a Soxhlet thimble and extract with 100-200 mL of the solvent for 12-24 hours [57] [54].
- Batchwise Extraction: Incubate the MIP with the solvent (approx. 10 mL per gram of MIP) on an orbital shaker or in an ultrasonic bath, refreshing the solvent every few hours until no template is detected in the eluent.
- Neutralization Wash: Wash the MIP with pure methanol to remove residual acetic acid.
- Final Wash and Drying: Perform a final rinse with deionized water (for aqueous applications) and dry the MIP under vacuum at an appropriate temperature.
Key Considerations: Monitor the extraction process by analyzing the washings spectrophotometrically or chromatographically until no template is detected. While effective, Soxhlet extraction uses large solvent volumes and extended time; alternative methods like pressurized liquid extraction or ultrasound-assisted extraction may offer more efficiency [54].
Protocol 3: Electrochemical Template Removal for MIP-Based Sensors
Principle: Applying a controlled electrochemical potential to MIPs synthesized directly on electrode surfaces can induce template desorption via redox reactions, offering an in-situ, chemical-free removal method [56].
Materials:
- MIP-coated electrode
- Electrochemical workstation (e.g., potentiostat)
- Appropriate electrolyte solution (e.g., phosphate buffer saline)
- Standard three-electrode cell (working, counter, and reference electrodes)
Procedure:
- Setup: Immerse the MIP-coated working electrode in an electrochemical cell containing the electrolyte solution.
- Conditioning: Perform an initial conditioning step by cycling the potential in a window where no faradaic processes occur to stabilize the system.
- Template Removal: Run multiple cycles of cyclic voltammetry (e.g., 20-50 cycles) in a predetermined potential window that facilitates the oxidation or reduction of the template-monomer complex, leading to template ejection.
- Verification: Monitor the voltammetric signal stabilization, which indicates the completion of the template removal process. The electrode is then ready for use or characterization.
Key Considerations: This method is particularly suited for electrochemical sensors and allows for electrode regeneration. It is simple and avoids the use of solvents, but its efficiency depends on the electroactivity of the template and the MIP matrix [56].
Quantitative Analysis of MIP Performance
Binding Isotherm Models for Characterizing Heterogeneity
The binding properties of MIPs are best described by models that account for site heterogeneity. The table below summarizes the key models used to quantify binding affinity and heterogeneity.
Table 1: Binding Isotherm Models for Characterizing MIP Heterogeneity
| Model Name | Mathematical Form | Key Parameters | Interpretation | Applicability |
|---|---|---|---|---|
| Langmuir (Homogeneous) | ( B = \frac{B{max} \cdot C}{Kd + C} ) | ( B{max} ): Total site capacity( Kd ): Dissociation constant | Assumes a single, uniform class of binding sites. Inaccurate for most MIPs [55]. | Homogeneous surfaces; not generally recommended for MIPs. |
| bi-Langmuir | ( B = \frac{B{max1} \cdot C}{K{d1} + C} + \frac{B{max2} \cdot C}{K{d2} + C} ) | ( B{max1}, B{max2} ): Capacities for two site types( K{d1}, K{d2} ): Affinities for two site types | Models two distinct classes of independent binding sites (e.g., specific and non-specific) [55]. | MIPs with two dominant, discrete site types. |
| Freundlich | ( B = a \cdot C^{m} ) | ( a ): Binding capacity( m ): Heterogeneity index (0 < m ≤ 1) | An empirical model. A lower m value indicates greater heterogeneity [55]. | Highly heterogeneous MIPs; useful over a limited concentration range. |
| Langmuir-Freundlich (SIPS) | ( B = \frac{B_{max} \cdot (a \cdot C)^{m}}{1 + (a \cdot C)^{m}} ) | ( B_{max} ): Total capacity( a ): Average affinity( m ): Heterogeneity index | A more general model. As m → 1, sites become homogeneous. Provides an affinity distribution [55]. | Recommended for most MIPs as it gives a quantitative measure of heterogeneity. |
Comparison of Template Removal Techniques
The efficiency of template removal is critical to minimize leaching. The following table compares different extraction methods.
Table 2: Comparison of Template Removal Techniques for MIPs
| Extraction Technique | Principle | Typical Conditions | Removal Efficiency / Yield | Advantages | Disadvantages |
|---|---|---|---|---|---|
| Soxhlet Extraction | Continuous extraction with refluxing solvent [54]. | Methanol/Acetic Acid (9:1), 12-24 hours | ~67-88% reported for theophylline MIPs [54] | Thorough, no filtration needed, affordable setup | Large solvent volume, long time, high temperature risk |
| Batch Solvent Washing | Incubation with solvent using agitation [57]. | Methanol/Acetic Acid (9:1), multiple washes | >90% achievable with sufficient solvent volumes [57] | Simple, scalable, works for thermolabile templates | Requires multiple steps and solvent monitoring |
| Electrochemical Removal | In-situ template desorption via applied potential [56]. | Cyclic Voltammetry, 20-50 cycles in buffer | Highly system-dependent; enables in-situ regeneration | Chemical-free, fast, suitable for sensor regeneration | Limited to electroactive templates/electrode MIPs |
| Ultrasound-Assisted | Solvent extraction enhanced by cavitation [54]. | Solvent in ultrasonic bath, <1 hour | Can achieve high yields more rapidly than Soxhlet [54] | Faster, potentially higher efficiency, lower temperature | Possible polymer degradation from intense ultrasound |
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 3: Key Reagent Solutions for MIP Synthesis and Evaluation
| Reagent / Material | Function / Role | Common Examples | Protocol Application |
|---|---|---|---|
| Functional Monomers | Provide interactive groups for template complexation [4]. | Methacrylic acid (MAA), Acrylic acid (AA), 4-Vinylpyridine (4-VP) | Fundamental to all MIP synthesis; choice dictates interaction strength. |
| Cross-linkers | Create a rigid polymer network to stabilize imprinted cavities [57]. | Ethylene glycol dimethacrylate (EGDMA), Trimethylolpropane trimethacrylate (TRIM) | Fundamental to all MIP synthesis; high cross-linking reduces swelling/leaching. |
| Surfactants | Suppress non-specific binding by masking external functional groups [1]. | Sodium dodecyl sulfate (SDS), Cetyl trimethyl ammonium bromide (CTAB) | Protocol 1: Post-synthesis modification of MIPs. |
| Porogenic Solvents | Dissolve polymerization components and create pore structure [57]. | Toluene, Acetonitrile, Chloroform | Fundamental to all MIP synthesis; affects cavity accessibility. |
| Extraction Solvents | Disrupt template-monomer interactions for template removal [56] [54]. | Methanol:Acetic Acid (9:1 v/v) | Protocol 2: Chemical extraction of the template. |
The following diagram summarizes the integrated strategies for addressing template leaching and binding site heterogeneity in MIP development.
Template leaching and binding site heterogeneity are interconnected challenges that fundamentally impact MIP performance. Addressing them requires an integrated strategy spanning rational pre-polymerization design, efficient and validated template removal, and post-synthesis modifications. The protocols and analytical methods detailed in this application note provide a structured framework for researchers to systematically produce more reliable and effective molecularly imprinted polymers. By employing computational screening to optimize monomer-template interactions, implementing rigorous template removal protocols while monitoring efficiency, utilizing surfactant modifications to minimize non-specific binding, and quantitatively characterizing the resulting binding site heterogeneity using appropriate models, scientists can significantly advance the application of MIPs in sensitive analytical detection, targeted drug delivery, and other advanced fields.
Strategies for Improving Biocompatibility and In Vivo Performance
The translational application of Molecularly Imprinted Polymers (MIPs) in biomedicine, particularly for drug delivery, biosensing, and tissue engineering, is critically dependent on their biocompatibility and performance within a living organism (in vivo). While MIPs offer the distinct advantage of being highly stable and customizable synthetic receptors, their journey from a laboratory material to a clinical tool is often hampered by challenges such as non-specific binding and unfavorable immune responses [1] [58]. This document outlines key strategies, grounded in recent scientific literature, to enhance the biocompatibility and in vivo efficacy of MIPs. The protocols and data presented herein are framed within the broader research objective of developing next-generation MIPs with minimized non-specific interactions for more precise and safer biomedical applications.
Core Strategies and Experimental Protocols
Two primary, interconnected approaches for enhancing MIP performance are the suppression of non-specific adsorption and the comprehensive evaluation of in vivo biodistribution and cytotoxicity.
Strategy I: Suppression of Non-Specific Adsorption via Surfactant Modification
Non-specific adsorption occurs when functional groups on the MIP's surface, located outside the specific imprinted cavities, interact with molecules other than the target analyte. This phenomenon severely compromises the selectivity and performance of MIPs, especially in complex biological fluids [1] [16].
Experimental Protocol: Electrostatic Modification of MIPs with Surfactants
This protocol details a method to mitigate non-specific binding by blocking external functional groups with surfactants, as demonstrated for the detection of sulfamethoxazole (SMX) [1] [16].
Materials
- Functional Monomers: Methacrylic acid (MAA) or 4-vinylpyridine (4-VP).
- Cross-linker: Ethylene glycol dimethacrylate (EGDMA).
- Template: Target molecule (e.g., Sulfamethoxazole, SMX).
- Initiator: 2,2'-Azobisisobutyronitrile (AIBN).
- Surfactants: Sodium dodecyl sulfate (SDS) and Cetyl trimethyl ammonium bromide (CTAB).
- Solvents: Dimethylformamide (DMF), Acetic acid, Methanol.
Procedure
- MIP Synthesis: Synthesize the MIP via a standard bulk polymerization method.
- Prepare a pre-polymerization mixture containing the template (e.g., SMX), functional monomer (MAA for a negatively charged polymer or 4-VP for a positively charged polymer), cross-linker (EGDMA), and initiator (AIBN) in a porogenic solvent (e.g., DMF).
- Purge the mixture with nitrogen to remove oxygen and initiate polymerization thermally or via UV light.
- After polymerization, grind the resulting monolith and sieve to obtain a desired particle size fraction.
- Template Removal: Wash the MIP particles extensively with a washing solvent (e.g., methanol:acetic acid, 9:1 v/v) to remove the template molecules until no template can be detected in the supernatant.
- Surfactant Modification: Incubate the template-free MIP particles with an aqueous solution of the selected surfactant.
- For a MIP synthesized with 4-VP (positively charged), use the anionic surfactant SDS.
- For a MIP synthesized with MAA (negatively charged), use the cationic surfactant CTAB.
- The electrostatic interaction between the surfactant and the external functional groups effectively "caps" them, preventing non-specific interactions.
- Validation: The success of the modification is validated by comparing the binding isotherms and adsorption kinetics of the target molecule on the MIP before and after surfactant treatment, and against a non-imprinted polymer (NIP). A successful modification shows a significant reduction in the NIP's binding capacity (non-specific adsorption) while largely preserving the MIP's specific binding [1].
- MIP Synthesis: Synthesize the MIP via a standard bulk polymerization method.
The following workflow diagram illustrates this experimental process:
Strategy II: Comprehensive In Vivo Biocompatibility and Biodistribution Assessment
Before MIPs can be deployed in clinical applications, a rigorous assessment of their behavior in a living system is mandatory. This involves evaluating their distribution throughout the body, clearance pathways, and potential cytotoxicity [59].
Experimental Protocol: Assessing In Vivo Biodistribution and Clearance of NanoMIPs
This protocol is adapted from a study investigating the in vivo fate of molecularly imprinted polymer nanoparticles (nanoMIPs) in a rodent model [59].
Materials
- NanoMIPs: Fluorescently labelled (e.g., with DiD dye) or radiolabelled nanoMIPs.
- Animal Model: Rats or mice (e.g., Sprague-Dawley rats).
- Administration Vehicles: Saline for intravenous (IV) injection; appropriate formulation for oral gavage.
- Analytical Instruments: IVIS imaging system, HPLC-MS, or a gamma counter depending on the label.
Procedure
- Dosing and Administration:
- Divide animals into experimental groups (e.g., IV vs. oral administration).
- Administer a defined dose of labelled nanoMIPs. For IV injection, a common dose is a blood concentration of 250 µg/mL [60].
- Blood Clearance Kinetics:
- Collect small blood samples (<1% of total blood volume) at multiple time points post-administration.
- Measure the concentration of nanoMIPs in the blood using a high-throughput quantitative method (e.g., fluorescence-based assay). This data is used to calculate the circulation half-life.
- Biodistribution Analysis:
- At predetermined time points, euthanize the animals and harvest major organs (e.g., heart, lungs, liver, spleen, kidneys, and brain).
- Quantitative Imaging: Use whole-organ IVIS imaging to qualitatively and quantitatively assess the distribution of the fluorescent signal.
- Cellular Analysis: Homogenize organs and use flow cytometry to identify which specific cell types have taken up the nanoMIPs (e.g., F4/80+ macrophages in the liver).
- Clearance Pathway Determination:
- Collect feces and urine over a defined period.
- Analyze the excreta for the presence of the nanoMIPs or their degradation products to determine the primary routes of clearance (renal vs. hepatic).
- Immunogenicity and Cytotoxicity:
- Monitor animals for signs of systemic toxicity.
- Collect blood for hematological and clinical chemistry analysis.
- Perform histological analysis (H&E staining) on tissues to assess local inflammation or damage.
- Dosing and Administration:
Table 1: Key Findings from a Representative In Vivo Study of NanoMIPs [59]
| Parameter | Finding | Implication for Biocompatibility |
|---|---|---|
| Biodistribution | Found in all harvested tissues, including the brain. | Demonstrates ability to cross biological barriers like the blood-brain barrier, useful for CNS targeting. |
| Clearance Route | Cleared via both feces and urine. | Indicates both renal and hepatic clearance mechanisms, suggesting no single-organ accumulation burden. |
| Cytotoxicity | Relatively low cytotoxicity observed. | Supports the safety profile of the tested nanoMIPs for further development. |
| Immunogenicity | MIPs specific for a cell surface protein showed moderate adjuvant properties; control MIPs did not. | Highlights that immunogenicity is template-dependent and must be evaluated on a case-by-case basis. |
The Scientist's Toolkit: Essential Research Reagents
The following table lists key materials and reagents essential for implementing the strategies described in this document.
Table 2: Key Reagents for Enhancing MIP Biocompatibility and Performance
| Reagent / Material | Function / Role | Specific Example |
|---|---|---|
| Sodium Dodecyl Sulfate (SDS) | Anionic surfactant used to cap positively charged external functional groups on MIPs to reduce non-specific binding. | Used to modify MIPs made with 4-vinylpyridine monomer [1]. |
| Cetyl Trimethyl Ammonium Bromide (CTAB) | Cationic surfactant used to cap negatively charged external functional groups on MIPs to reduce non-specific binding. | Used to modify MIPs made with methacrylic acid monomer [1]. |
| Polyethylene Glycol (PEG) | Polymer used to create a hydrophilic "stealth" coating on nanoparticles, prolonging blood circulation time by reducing phagocytic clearance. | PACE-PEG NPs exhibited prolonged blood circulation compared to unPEGylated NPs [60]. |
| Methacrylic Acid (MAA) | A common functional monomer for creating non-covalent imprinting sites via hydrogen bonding and ionic interactions. | Used as a monomer for imprinting sulfamethoxazole [1]. |
| Ethylene Glycol Dimethacrylate (EGDMA) | A cross-linker that provides mechanical stability to the MIP matrix and maintains the structure of the imprinted cavities. | Commonly used cross-linker in both bulk and surface imprinting protocols [1] [15]. |
| Fluorescent Dyes (e.g., DiD) | Lipophilic tracers for labeling nanoparticles to enable tracking and quantification in in vivo biodistribution studies. | Used to label PACE NPs for in vivo imaging and flow cytometry analysis [60]. |
The strategic enhancement of MIPs for in vivo applications is a multi-faceted endeavor. The integration of surface modification techniques, such as surfactant treatment to minimize non-specific binding, with a rigorous in vivo assessment protocol to understand pharmacokinetics and safety, provides a robust framework for advancing MIP technology. By systematically applying these strategies, researchers can develop more reliable, selective, and biocompatible MIP-based systems, thereby accelerating their translation into real-world diagnostic and therapeutic applications.
Evaluating MIP Selectivity: Methods and Comparative Material Analysis
Molecularly imprinted polymers (MIPs) are synthetic, highly cross-linked polymers with tailor-made recognition sites complementary to a target analyte in terms of shape, size, and functional groups [53]. Their efficacy, however, is often compromised by non-specific adsorption on non-imprinted polymer (NIP) regions, which reduces selectivity [16] [15]. This application note details three fundamental validation techniques—batch rebinding assays, chromatographic analysis, and Scatchard plots—essential for characterizing MIP performance within a research context focused on reducing non-specific binding. These protocols provide a framework for researchers and drug development professionals to quantitatively assess the binding characteristics and specificity of novel MIPs.
Experimental Protocols
Batch Rebinding Assays
Batch rebinding is a fundamental method for evaluating the binding capacity and affinity of MIPs in a liquid phase [61].
- Principle: The method involves incubating a known amount of MIP particles with a solution of the target analyte. After reaching equilibrium, the concentration of free analyte in the supernatant is measured to calculate the amount bound to the polymer [61].
- Procedure:
- Polymer Preparation: Synthesize MIP and corresponding NIP. Grind the bulk polymer and sieve to obtain particles of a defined size range (e.g., 100–300 μm) [61]. Extract the template molecules thoroughly, verified by UV spectroscopy of the supernatant until no template is detected [57] [61].
- Equilibration: Accurately weigh a fixed amount (e.g., 10-50 mg) of MIP or NIP into a vial. Add a known volume of a standard solution of the target analyte at a specific concentration. Seal the vials and agitate them in a thermostated shaker for a predetermined equilibration time (e.g., 12-24 hours) [61].
- Separation and Analysis: Centrifuge the mixture or separate the particles using filtration. Measure the concentration of the unbound analyte in the supernatant using a suitable technique such as UV-Vis spectrophotometry [57] or HPLC.
- Data Calculation: The amount of analyte bound to the polymer (Q) is calculated using the formula: ( Q = \frac{(Ci - Cf) \times V}{m} ), where ( Ci ) and ( Cf ) are the initial and final concentrations of the analyte, respectively, ( V ) is the volume of the solution, and ( m ) is the mass of the polymer.
The workflow for a batch rebinding assay is systematically outlined in the diagram below.
Chromatographic Analysis
Chromatographic evaluation assesses the MIP's ability to function as a selective stationary phase, directly probing its recognition properties [62].
- Principle: The MIP particles are packed into a chromatographic column. The retention characteristics of the template molecule on the MIP column are compared to those on a NIP column or for structural analogs, providing a direct measure of selectivity [62].
- Procedure:
- Column Packing: Ground and sieved MIP particles (e.g., <25 μm) are slurry-packed into a suitable HPLC column (e.g., stainless steel) [62]. A control column packed with NIP should be prepared identically.
- Chromatographic Evaluation: Prepare solutions of the template and its structural analogs. Inject these samples onto the MIP and NIP columns.
- Mobile Phase: Use an optimized mobile phase that supports the molecular recognition process, typically involving a weak solvent (like an aqueous buffer) to promote binding and a strong solvent (like acetonitrile or methanol) for elution [62]. Isocratic or gradient elution can be employed.
- Data Analysis: Measure the retention time (t₍ᵣ₎) and capacity factor (k') for each analyte. The imprinting factor (IF) is a key metric of selectivity, calculated as ( IF = \frac{k'{MIP}}{k'{NIP}} ), where ( k'{MIP} ) and ( k'{NIP} ) are the capacity factors on the MIP and NIP columns, respectively [62]. A higher IF indicates superior specificity.
Scatchard Analysis
The Scatchard plot is used to analyze binding data from batch experiments to estimate the binding parameters and heterogeneity of the imprinted sites [61] [63].
- Principle: This model analyzes the equilibrium binding data to estimate the affinity and population density of different classes of binding sites within the MIP.
- Procedure:
- Data Collection: Perform batch rebinding assays as described in Section 2.1 across a wide range of initial analyte concentrations.
- Plot Construction: Plot ( \frac{Q}{[Cf]} ) on the y-axis versus ( Q ) on the x-axis, where ( Q ) is the bound analyte concentration and ( [Cf] ) is the free analyte concentration at equilibrium.
- Data Fitting and Interpretation: Non-linear Scatchard plots are common for MIPs, indicating site heterogeneity [61] [63]. The data can be fit to a multi-site model (e.g., a two-site model). The slope of a linear region is ( -KA ) (association constant), and the x-intercept gives the apparent maximum binding capacity (( Q{max} )) for that site class.
The following diagram illustrates the logical process for interpreting a Scatchard plot, which is critical for understanding MIP binding heterogeneity.
Data Presentation and Analysis
Table 1: Key performance metrics obtained from MIP validation techniques.
| Technique | Primary Metrics | Interpretation | Reported Example Values |
|---|---|---|---|
| Batch Rebinding | Binding Capacity (Q), Imprinting Factor (IF = QMIP/QNIP) | Measures total uptake and specificity. | IF of 3.75 for Methyl Red MIP [57]; IF of 3.38 for a 2,4-D MIP with reduced non-specific binding [15]. |
| Chromatographic Analysis | Retention Time (tR), Capacity Factor (k'), Imprinting Factor (IF = k'MIP/k'NIP) | Measures separation efficiency and chiral resolution. | Corrected selectivity of 6.8 for pentamidine vs. benzamidine [62]. |
| Scatchard Analysis | Association Constant (KA), Maximum Binding Capacity (Qmax) | Quantifies binding affinity and site heterogeneity. | A 2-FAL MIP showed three distinct site classes with different affinities [61]. |
Research Reagent Solutions
Table 2: Essential materials and reagents for MIP synthesis and validation.
| Reagent/Material | Function | Common Examples |
|---|---|---|
| Functional Monomer | Provides interactive groups for template complexation via non-covalent bonds. | Methacrylic acid (MAA) [57] [61], Vinylpyridine [62]. |
| Cross-linking Monomer | Creates a rigid polymer network to stabilize the imprinted cavities. | Ethylene glycol dimethacrylate (EGDMA) [57] [15], Divinyl benzene (DVB) [61]. |
| Initiator | Starts the free-radical polymerization reaction. | 2,2'-Azobisisobutyronitrile (AIBN) [57] [61]. |
| Porogenic Solvent | Dissolves all components and creates pore structure during polymerization. | Toluene [57], Acetonitrile. |
| Template Molecule | Target molecule around which the complementary cavity is formed. | Methyl Red [57], 2-Furaldehyde [61], Pharmaceuticals [62]. |
| Washing Solvent | Extracts the template molecule from the polymerized network. | Methanol:Acetic acid (9:1 v/v) [57], Ethanol [61]. |
Molecularly imprinted polymers (MIPs) are synthetic materials designed with specific recognition sites complementary to target molecules in shape, size, and functional groups [1]. Despite their significant advantages over biological recognition elements, conventional MIPs often face challenges including slow mass transfer, incomplete template removal, and limited accessibility to binding sites [64]. Surface imprinting technology has emerged as a powerful strategy to overcome these limitations by creating recognition sites on the surface of solid supports, significantly enhancing binding kinetics and site accessibility [64] [65].
The selection of an appropriate support material is crucial for determining the final performance of surface-imprinted MIPs. This application note provides a systematic comparison of three advanced support materials: zeolite Y, silica aerogel, and MIL-101(Cr) (a metal-organic framework), for creating high-performance MIPs targeting fuel ether oxygenates in drinking water [64]. The comparative data and protocols presented herein aim to guide researchers in selecting optimal support materials for specific applications, particularly within the broader context of developing MIPs with reduced non-specific binding.
Comparative Performance Data
The following tables summarize the key characteristics and performance metrics of MIPs synthesized on the three different support materials, as determined through a comprehensive comparative study [64].
Table 1: Support Material Properties and Characterization Data
| Parameter | Zeolite Y-MMIP | Silica Aerogel-MMIP | MIL-101(Cr)-MMIP |
|---|---|---|---|
| BET Surface Area (m²/g) | 59.4 | Not Specified | Not Specified |
| Total Pore Volume (cm³/g) | 0.2034 | Not Specified | Not Specified |
| Average Pore Size (nm) | 13.7 | Not Specified | Not Specified |
| Key Support Advantages | Large surface area, uniform pore size, chemical/thermal stability [64] | High surface area, low density, high porosity [64] | High specific surface area, mechanical strength, tunable porosity [64] [66] |
Table 2: Analytical Performance for Fuel Ether Oxygenate Extraction
| Analyte | LOD (μg L⁻¹) | LOQ (μg L⁻¹) | Linear Range (μg L⁻¹) | Adsorption Capacity |
|---|---|---|---|---|
| Methyl tert-butyl ether (MTBE) | 0.64 | 2.1 | 1 - 100 | Not Specified |
| Ethyl tert-butyl ether (ETBE) | 0.40 | 1.3 | 1 - 100 | Not Specified |
| tert-Butyl formate (TBF) | 0.34 | 1.1 | 1 - 100 | Not Specified |
| Optimal Adsorbent | Zeolite Y-MMIP (selected for all analytes) [64] | |||
| Optimal Conditions | Adsorbent: 40 mg; pH: 7.7; Absorption time: 24.8 min [64] |
Abbreviations: LOD = Limit of Detection; LOQ = Limit of Quantification.
Experimental Protocols
Support Preparation and Magnetization
The protocol begins with the preparation and magnetization of the candidate support materials to facilitate easy separation during the dispersive solid-phase extraction (DSPE) process [64].
Materials Functionalization
- Silica aerogel, Zeolite Y, and MIL-101(Cr) are commercially sourced [64].
- Magnetization: The surfaces of all three materials are modified to impart magnetic properties. This typically involves in-situ precipitation of iron oxides (e.g., Fe₃O₄) onto the support surface or layer-by-layer self-assembly of magnetic nanoparticles [64] [67].
- Surface Activation: The magnetized supports are further functionalized with vinyltriethoxysilane (VTES). VTES acts as a coupling agent, introducing vinyl groups (-CH=CH₂) onto the support surface. These groups participate in the subsequent polymerization reaction, anchoring the imprinted polymer layer to the support [64].
Surface Molecular Imprinting Polymerization
This critical step creates the selective recognition layer on the functionalized and magnetized supports.
Pre-polymerization Complex Formation
- Template: Fuel ether oxygenates (MTBE, ETBE, TBF) or a structural analog.
- Functional Monomer: Methacrylic acid (MAA) is commonly used. The monomer is selected for its ability to form non-covalent interactions (e.g., hydrogen bonds) with the template molecule [64] [66].
- Procedure: The template and functional monomer are dissolved in a suitable porogen solvent (e.g., toluene or acetonitrile) and allowed to equilibrate. During this stage, the monomer surrounds the template, forming a pre-arranged complex [66].
Polymerization
- Cross-linker: Ethylene glycol dimethacrylate (EGDMA) is added to create a rigid, three-dimensional polymer network [64] [66].
- Initiator: 2,2'-Azobisisobutyronitrile (AIBN) is added to initiate free-radical polymerization [64] [66].
- Reaction: The functionalized magnetic support from Step 3.1.1 is dispersed in the pre-polymerization mixture. The polymerization is carried out under an inert nitrogen atmosphere at 60°C for 24 hours [64] [66]. The vinyl groups on the support surface covalently integrate with the growing polymer chains.
Template Removal
- After polymerization, the template molecules are removed from the polymer matrix by Soxhlet extraction using a mixture of methanol and acetic acid (9:1, v/v) [64] [66]. This critical step liberates the specific binding cavities, making them available for the target analyte.
DSPE Optimization and Validation
The optimal MIP (Zeolite-Y-MMIP) is evaluated for extracting target analytes from water samples.
Optimization of Extraction
- Critical parameters (adsorbent amount, pH, extraction time) are optimized using Central Composite Design-Response Surface Methodology (CCD-RSM) to understand interaction effects and identify optimal conditions [64].
- Optimal Conditions for Fuel Ether Oxygenates: 40 mg of Zeolite-Y-MMIP adsorbent, pH of 7.7, and an absorption time of 24.8 minutes [64].
Extraction Procedure
- Add 40 mg of the optimal MIP to the water sample.
- Adjust the sample pH to 7.7.
- Agitate the mixture for 25 minutes to allow analyte binding.
- Separate the MIP sorbent using an external magnet.
- Elute the bound analytes with a small volume of organic solvent (e.g., methanol).
- Analyze the eluent using gas chromatography or another suitable analytical technique [64].
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Reagents for MIP Synthesis and Evaluation
| Reagent | Function/Application | Examples & Notes |
|---|---|---|
| Support Materials | Provides a high-surface-area scaffold for surface imprinting. | Zeolite Y, Silica Aerogel, MIL-101(Cr) [64]. |
| Functional Monomers | Interacts with the template to form a pre-polymerization complex. | Methacrylic acid (MAA), 4-Vinylpyridine (4-VP) [64] [65]. |
| Cross-linkers | Creates a rigid polymer network to stabilize the imprinted cavities. | Ethylene glycol dimethacrylate (EGDMA), Divinylbenzene (DVB) [64] [65]. |
| Initiators | Initiates the free-radical polymerization reaction. | 2,2'-Azobisisobutyronitrile (AIBN) [64] [66]. |
| Porogen Solvents | Dissolves all components and creates pore structure during polymerization. | Toluene, Acetonitrile, Dimethylformamide (DMF) [64] [66]. |
| Surfactants (for NSA Reduction) | Electrostatically blocks external functional groups to minimize non-specific binding. | Sodium dodecyl sulfate (SDS), Cetyl trimethyl ammonium bromide (CTAB) [1] [16]. |
Workflow and Signaling Visualizations
Experimental Workflow for MIP Synthesis and Application
The following diagram illustrates the comprehensive workflow for the synthesis of surface-imprinted MIPs on different supports and their application in dispersive solid-phase extraction.
Mechanism of Non-Specific Adsorption Suppression
A key challenge in MIP development is non-specific adsorption (NSA), which reduces selectivity. The following diagram illustrates an innovative strategy using surfactants to suppress NSA.
This comparative study demonstrates that the choice of support material significantly influences the performance of molecularly imprinted polymers. For the extraction of fuel ether oxygenates from water, Zeolite Y-based MIPs outperformed those built on silica aerogel and MIL-101(Cr), showing superior selectivity and the lowest detection limits for the target analytes [64]. The optimal DSPE protocol utilizing 40 mg of Zeolite-Y-MMIP at pH 7.7 with a 25-minute extraction time provides a robust method for monitoring these contaminants in drinking water at trace levels.
The integration of surface imprinting with advanced support materials like Zeolite Y, combined with strategies to mitigate non-specific binding such as surfactant modification, represents a significant advancement in the design of highly selective MIPs. These protocols and findings provide a solid foundation for researchers developing sensitive and selective extraction materials for environmental, pharmaceutical, and biomedical analysis.
Molecularly imprinted polymers (MIPs) are synthetic materials designed with specific recognition sites for target molecules, functioning as "plastic antibodies" [1] [5]. The performance of these polymers is quantitatively assessed through three core metrics: the imprinting factor (IF), which measures the effectiveness of the imprinting process; binding affinity (Kd), which quantifies the strength of the template-MIP interaction; and selectivity coefficients, which evaluate the MIP's ability to discriminate the target from interferents [68] [5]. For research focused on reducing non-specific binding—a prevalent issue that compromises MIP performance and application reliability [1]—a rigorous and accurate understanding of these parameters is indispensable. This document provides detailed application notes and protocols for the quantitative analysis of these critical performance metrics, supporting the development of high-fidelity MIPs.
Core Performance Metrics and Quantitative Data
The following table summarizes the key performance metrics, their definitions, and representative values from recent literature, providing a benchmark for evaluating MIP performance.
Table 1: Core Performance Metrics for Molecularly Imprinted Polymers
| Metric | Definition & Purpose | Representative Values (from Literature) | Interpretation Guidelines |
|---|---|---|---|
| Imprinting Factor (IF) | A measure of the specificity conferred by the imprinting process. Calculated as IF = Binding to MIP / Binding to NIP [68] [5]. | IF > 1.3 for a high-performance EPMC MIP [68]; IF = 5.57 for a magnetic β-CD-MIP for PFOA [69]. | IF > 1 indicates successful imprinting. Higher values denote greater specificity and more effective cavity formation. |
| Binding Affinity (Kd) | The equilibrium dissociation constant, quantifying the strength of the template-MIP interaction. A lower Kd indicates higher affinity [68] [44]. | KF = 0.081 mg/g for a high-affinity MIP (B2) binding EPMC, based on Freundlich isotherm analysis [68]. | Lower Kd values signify tighter binding. Isotherm models (Freundlich, Langmuir) are used to derive affinity parameters. |
| Selectivity Coefficient (k) | Evaluates the MIP's ability to discriminate between the target analyte and structural analogues. k = IFtarget / IFcompetitor [68] [1]. | High discrimination of EPMC over structural analogues reported [68]; Surfactant-modified MIPs showed elimination of non-specific adsorption [1]. | k >> 1 indicates high selectivity for the target over the competitor. Values near 1 suggest poor discrimination. |
Experimental Protocols for Performance Evaluation
Protocol 1: Batch Rebinding Studies for IF and Kd
This fundamental protocol is used to determine the imprinting factor and binding affinity of a MIP.
3.1.1 Research Reagent Solutions
Table 2: Essential Materials for Batch Rebinding Experiments
| Reagent/Material | Function/Explanation | Example from Literature |
|---|---|---|
| MIP and NIP | The molecularly imprinted polymer and non-imprinted control are used in parallel to quantify specific vs. non-specific binding. | Synthesized via bulk polymerization with MAA monomer [68]. |
| Template/Analyte | The target molecule for which the MIP was synthesized. | Ethyl p-methoxycinnamate (EPMC) [68]; Sulfamethoxazole (SMX) [1]. |
| Structural Analogues | Compounds similar to the template; used to assess binding selectivity. | Sulfadiazine, Sulfamerazine for a Sulfamethoxazole MIP [1]. |
| Porogenic Solvent | The solvent used during polymerization; can significantly influence binding affinity and should be used in rebinding studies. | Chloroform, n-hexane [68]. |
| UV-Vis Spectrophotometer / HPLC | Analytical instrument to quantify the concentration of unbound analyte in solution after interaction with the polymer. | Used for quantification in adsorption studies [68] [1]. |
3.1.2 Methodology
- Preparation of Stock Solutions: Prepare a known concentration of the template (e.g., 100 µg/mL) in an appropriate solvent (e.g., the porogen used in synthesis).
- Equilibrium Binding: Weigh equal amounts (e.g., 10.0 mg) of MIP and NIP into separate vials. Add a fixed volume (e.g., 5.0 mL) of the template stock solution to each vial. Seal the vials and incubate with agitation (e.g., on a shaker) at a constant temperature until equilibrium is reached (typically 12-24 hours) [68] [5].
- Separation: Centrifuge the vials or use filtration to separate the polymer particles from the supernatant.
- Quantification: Analyze the concentration of the unbound template in the supernatant using a calibrated method such as UV-Vis spectrophotometry or HPLC [1].
- Data Calculation:
- Calculate the amount of template bound to the polymer (Q) using the formula: ( Q = \frac{(Ci - Cf) \times V}{m} ), where ( Ci ) and ( Cf ) are the initial and final concentrations, V is the solution volume, and m is the polymer mass.
- The Imprinting Factor (IF) is calculated as: ( IF = \frac{Q{MIP}}{Q{NIP}} ).
- Isotherm Modeling: Repeat steps 1-5 using a range of initial template concentrations. Plot Q vs. Cf (the adsorption isotherm) and fit the data to models like the Freundlich or Langmuir isotherm to derive binding affinity parameters (e.g., KF) [68].
The logical workflow for this protocol, from preparation to data analysis, is outlined below.
Protocol 2: Selectivity Assessment
This protocol evaluates the MIP's specificity by testing its binding towards structural analogues of the template.
3.2.1 Methodology
- Competitor Selection: Identify and obtain one or more compounds that are structurally related to the template molecule.
- Parallel Binding Experiments: Perform the batch rebinding protocol (Protocol 1, steps 2-5) separately for the template and each competitor molecule using both the MIP and NIP.
- Data Analysis and Calculation:
- Calculate the imprinting factor for the template (IFT) and for each competitor (IFC).
- Calculate the Selectivity Coefficient (k) for each competitor: ( k = \frac{IFT}{IFC} ).
- A selectivity coefficient significantly greater than 1 indicates that the MIP preferentially binds the target template over the competitor, confirming the specificity of the imprinted cavities [68] [1].
Advanced Strategies to Suppress Non-Specific Binding
A major challenge in MIP development is non-specific adsorption to generic sites outside the imprinted cavities, which elevates NIP binding and lowers the IF [1]. The following advanced strategies have proven effective in mitigating this issue, directly enhancing the measured performance metrics.
Table 3: Strategies for Reducing Non-Specific Binding in MIPs
| Strategy | Mechanism of Action | Experimental Support & Outcome |
|---|---|---|
| Electrostatic Modification with Surfactants | Surfactants (e.g., SDS, CTAB) bind to and block external functional groups on the polymer backbone responsible for non-specific interactions [1]. | Modification of MIPs with SDS/CTAB effectively eliminated non-specific adsorption in Sulfamethoxazole MIPs, significantly improving selectivity [1]. |
| Surface Imprinting | Confines the imprinted sites to the surface of a support material (e.g., magnetic nanoparticles, MOFs), preventing template embedding and improving site accessibility [69] [70]. | Magnetic MIPs synthesized via surface imprinting showed high affinity and easy template removal, leading to high imprinting factors (e.g., 5.57) [69] [70]. |
| Computational Monomer Selection | Uses molecular modeling to screen functional monomers for optimal binding energy with the template before synthesis, promoting the formation of stable, specific complexes [44]. | Protocols using automated screening of monomer libraries enable the rational design of MIPs with high binding affinity and reduced reliance on non-specific interactions [44]. |
| Use of "Green" Biomass Components | Incorporates natural polymers (e.g., β-cyclodextrin, chitosan) that offer specific, well-defined interaction modes, reducing random, non-specific binding [71] [69]. | β-cyclodextrin used as a functional monomer facilitates specific inclusion complex formation, as demonstrated in a MIP for PFOA [69]. |
The relationship between these strategies and the core performance metrics is illustrated in the following causal pathway diagram.
The rigorous characterization of MIPs through imprinting factor, binding affinity, and selectivity coefficients is non-negotiable for advancing research aimed at reducing non-specific binding. The protocols outlined herein provide a standardized framework for obtaining these critical performance metrics. By integrating advanced strategies—such as surfactant modification, surface imprinting, and computational design—researchers can systematically engineer next-generation MIPs with enhanced specificity and performance. This approach is fundamental for developing reliable MIP-based applications in drug development, diagnostic sensing, and environmental analysis.
Comparative Analysis: MIPs vs. Natural Antibodies
Molecularly Imprinted Polymers (MIPs) are synthetic biomimetic receptors engineered to recognize specific target molecules with antibody-like specificity. Often termed "plastic antibodies," they are created through a polymerization process around a template target molecule, which, once removed, leaves behind complementary cavities [1]. The following table summarizes the key advantages of MIPs over natural antibodies, making them attractive for research and development.
Table 1: Key Advantages of MIPs over Natural Antibodies
| Parameter | Molecularly Imprinted Polymers (MIPs) | Natural Antibodies | Experimental Evidence & Notes |
|---|---|---|---|
| Stability | High stability under harsh conditions (extreme pH, organic solvents, high temperature/pressure) [19] [39]. Retain recognition properties for several years in dry, room-temperature storage [39]. | Sensitive to denaturation; require controlled, often refrigerated, storage conditions [72]. | Stability against degradation is critical for applications in complex biological or environmental matrices [37]. |
| Production Cost | Cost-effective and easy to prepare [39] [40]. Use readily available acrylic/methacrylic monomers and polymerization techniques. | Very expensive to produce and purify [72]. | Cost-effectiveness is a significant driver for exploring MIPs as substitutes in diagnostics and therapeutics [72]. |
| Reusability | Excellent reusability over multiple binding and washing cycles without significant loss of performance [37] [40]. | Typically single-use or limited reusability due to irreversible denaturation. | A high degree of cross-linking in MIPs ensures structural integrity during repeated use [37]. |
| Development Time | Relatively rapid synthesis and optimization, especially with modern computational approaches [19]. | Lengthy development process involving biological systems (e.g., animal immunization or phage display). | Solid-phase synthesis and computer-aided design are accelerating MIP development [19] [39]. |
Experimental Protocols for MIP Evaluation
Protocol: Solid-Phase Synthesis of High-Affinity NanoMIPs
This protocol outlines the synthesis of MIP nanoparticles (MIP NPs) using a solid-phase approach, which yields products with high affinity and selectivity and minimizes template residue issues [39].
1. Immobilization of Template:
- Covalently immobilize the target template molecule (e.g., a protein biomarker or a small-molecule drug) onto a solid support, such as functionalized glass beads or magnetic particles.
2. Polymerization:
- Prepare a pre-polymerization mixture containing the functional monomer (e.g., methacrylic acid for hydrogen bonding or 4-vinylpyridine for acidic templates), cross-linker (e.g., ethylene glycol dimethacrylate, EGDMA), initiator (e.g., azobisisobutyronitrile, AIBN), and a porogenic solvent (e.g., toluene or acetonitrile).
- Incubate the mixture with the template-immobilized beads to allow monomers to assemble around the template.
- Initiate polymerization thermally or photochemically.
3. Template Removal and Harvesting:
- Wash the polymer-coated beads extensively with a mild acid or organic solvent to remove the template molecule. In solid-phase synthesis, this step efficiently extracts the template, leaving behind specific cavities.
- Recover the resulting MIP NPs from the solution. The nanoparticles exhibit high binding affinity and are virtually free of template residues [39].
Protocol: Suppression of Non-Specific Adsorption via Surfactant Modification
Non-specific adsorption, caused by functional groups located outside the imprinted cavities, is a major challenge. This protocol details an electrostatic modification strategy to mitigate this issue [1].
1. MIP Synthesis:
- Synthesize MIPs via a suitable method (e.g., bulk or precipitation polymerization) using the target analyte (e.g., Sulfamethoxazole, SMX) as the template.
2. Surfactant Modification:
- For a MIP with a positively charged surface (e.g., poly(4-vinylpyridine)), incubate with the anionic surfactant Sodium Dodecyl Sulfate (SDS).
- For a MIP with a negatively charged surface (e.g., polymethacrylic acid), incubate with the cationic surfactant Cetyl Trimethyl Ammonium Bromide (CTAB).
- The surfactants will electrostatically react with and block the external functional groups responsible for non-specific binding, while the functional groups inside the specific cavities remain accessible.
3. Validation:
- Analyze binding isotherms of the target molecule on the surfactant-modified MIPs (MIP±-SDS/CTAB) and compare them to unmodified MIPs and Non-Imprinted Polymers (NIPs).
- Successful modification is indicated by a significant reduction in the adsorption capacity of the NIPs and the elimination of non-specific adsorption in the MIPs, while the specific adsorption capacity of the MIPs is retained [1].
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Reagents for MIP Development and Analysis
| Reagent / Material | Function / Application | Specific Examples |
|---|---|---|
| Functional Monomers | Form interactions with the template molecule to create specific binding sites. | Methacrylic acid (MAA), 4-Vinylpyridine, Acrylamide [37]. |
| Cross-Linkers | Provide rigidity and stability to the polymer matrix, "freezing" the cavities. | Ethylene glycol dimethacrylate (EGDMA), Divinylbenzene (DVB) [39] [37]. |
| Template Molecules | Serve as the "mold" for creating specific recognition cavities. | Drugs (Sulfamethoxazole), pesticides, biomarkers, proteins [37] [73]. |
| Surfactants | Modify MIP surface to suppress non-specific adsorption. | Sodium Dodecyl Sulfate (SDS), Cetyl Trimethyl Ammonium Bromide (CTAB) [1]. |
| Porogenic Solvents | Dissolve polymerization components and create pore structure. | Toluene, Acetonitrile, Chloroform [37]. |
Workflow Visualization
The following diagram illustrates the core process of creating and applying MIPs, integrating the key steps from synthesis to application and highlighting the strategic step for suppressing non-specific binding.
MIP Development and Application Workflow
MIPs present a compelling alternative to natural antibodies, demonstrating superior stability, cost-effectiveness, and reusability. These advantages are particularly valuable for applications in harsh environments, point-of-care diagnostics, and processes requiring multiple use cycles. Continued research and standardized protocols, such as those for suppressing non-specific binding, are paving the way for the broader commercial translation and acceptance of MIPs in pharmaceutical and biomedical analysis [73] [1].
Conclusion
The pursuit of molecularly imprinted polymers with minimal non-specific binding is paramount for their transition from robust synthetic materials to reliable tools in critical biomedical and clinical applications. Key takeaways include the proven efficacy of surfactant modification and surface imprinting to block non-specific sites, the necessity of systematic synthesis optimization, and the importance of using rigorous, application-relevant validation methods. Future progress hinges on an interdisciplinary approach that merges rational design powered by computational modeling with the development of novel, biocompatible materials. Promising directions include the refinement of biodegradable MIPs for drug delivery, the creation of highly sensitive MIP-nanozyme hybrids for point-of-care diagnostics, and the standardization of synthesis protocols to ensure reproducibility and foster wider commercial adoption.
References
- 1. Innovative approaches to suppress non-specific adsorption ... [https://www.sciencedirect.com/science/article/abs/pii/S0956566324000563]
- 2. Improving MIP-based electrochemical sensors by ... [https://www.sciencedirect.com/science/article/abs/pii/S0026265X25024634]
- 3. The Selectivity of Molecularly Imprinted Polymers - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC8198654/]
- 4. Computational and Experimental Comparison of ... [https://www.mdpi.com/1420-3049/29/17/4236]
- 5. Molecularly Imprinted Polymers: Present and Future ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3189760/]
- 6. From Antibodies to Molecularly Imprinted Polymers (MIPs) ... [https://www.mdpi.com/2079-4983/13/1/12]
- 7. MIPs and Aptamers for Recognition of Proteins in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5039654/]
- 8. 4 Ways to Reduce Non-Specific Binding in SPR Experiments [https://nicoyalife.com/blog/4-ways-reduce-non-specific-binding-spr/]
- 9. 8. SEM: Scanning Electron Microscopy [https://ncstate.pressbooks.pub/advancesinpolymerscience/chapter/sem-scanning-electron-microscopy-characterization-of-polymers/]
- 10. How can Polymers be Characterized Using SEM? [https://www.azom.com/article.aspx?ArticleID=23052]
- 11. Synthesis and Characterization of a New Molecularly ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10223826/]
- 12. The Infrared Spectra of Polymers, Part I: Introduction [https://www.spectroscopyonline.com/view/the-infrared-spectra-of-polymers-part-i-introduction]
- 13. Structural, spectroscopic, morphological and optical ... [https://www.nature.com/articles/s41598-025-10486-0]
- 14. A Review on Molecularly Imprinted Polymers Preparation ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8398116/]
- 15. Improving Surface Imprinting Effect by Reducing ... [https://www.mdpi.com/2227-9040/9/3/43]
- 16. Innovative approaches to suppress non-specific adsorption ... [https://pubmed.ncbi.nlm.nih.gov/38266615/]
- 17. Factors Affecting Preparation of Molecularly Imprinted Polymer and Methods on Finding Template-Monomer Interaction as the Key of Selective Properties of the Materials - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC8470890/]
- 18. Characterization of the Binding Properties of Molecularly Imprinted Polymers - PubMed [https://pubmed.ncbi.nlm.nih.gov/25796622/]
- 19. Molecularly Imprinted Polymers for Biomarker Detection [https://www.sciencedirect.com/science/article/pii/S0165993625003553]
- 20. Molecularly imprinted polymers by the surface ... [https://www.sciencedirect.com/science/article/abs/pii/S0014305720319480]
- 21. Strategies for Molecular Imprinting and the Evolution of MIP ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6940816/]
- 22. Molecularly imprinted polymers (MIPs): emerging biomaterials ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10182667/]
- 23. A comparison of the performance of molecularly imprinted ... [https://www.nature.com/articles/srep37638]
- 24. Recent advances in molecularly imprinted polymer-based ... [https://www.sciencedirect.com/science/article/pii/S0956566324000216]
- 25. Molecularly imprinted polymer-integrated nanozymes for ... [https://pubs.rsc.org/en/content/articlehtml/2025/tb/d5tb01416f?page=search]
- 26. Effect of Surfactants on the Binding Properties of a Molecularly ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9741244/]
- 27. Development of molecularly imprinted polymers for the ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12642357/]
- 28. A computational approach to study functional monomer ... [https://pubmed.ncbi.nlm.nih.gov/28444792/]
- 29. Computational modelling and optimization studies of ... [https://www.sciencedirect.com/science/article/abs/pii/S1093326324000159]
- 30. A computational approach to studying monomer selectivity ... [https://link.springer.com/article/10.1007/s00894-008-0437-2]
- 31. Interpretable machine learning for solvent prediction and ... [https://www.sciencedirect.com/science/article/pii/S1385894725102404]
- 32. Machine learning solves complex solvent selection challenge [https://engineering.wisc.edu/blog/machine-learning-solves-complex-solvent-selection-challenge/]
- 33. Machine Learning-Driven Solvent Screening for Biobased ... [https://www.ornl.gov/publication/machine-learning-driven-solvent-screening-biobased-23-butanediol-extraction]
- 34. Development of flexible regenerable lactate-specific ... [https://pubmed.ncbi.nlm.nih.gov/40479783/]
- 35. Automatic reactor for solid-phase synthesis of molecularly ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4693954/]
- 36. Universal peptide synthesis via solid-phase methods fused ... [https://www.nature.com/articles/s41467-025-62344-2]
- 37. Small Toxic Molecule Detection and Elimination Using ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12190631/]
- 38. Recent Advances in Molecularly Imprinted Polymers and ... [https://www.mdpi.com/2073-4360/15/21/4199]
- 39. Advances in Molecularly Imprinted Polymers as Drug Delivery ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8231147/]
- 40. Molecularly Imprinted Polymer Nanoparticles for ... [https://www.mdpi.com/2073-4360/17/17/2283]
- 41. Recent molecularly imprinted polymers applications in ... [https://link.springer.com/article/10.1007/s11696-022-02488-3]
- 42. A novel method for selective adsorption of toxic H₂S gas ... [https://www.nature.com/articles/s41598-025-16898-2]
- 43. Development and evaluation of optimized molecularly ... [https://www.sciencedirect.com/science/article/pii/S1570180825000545]
- 44. A Protocol for the Computational Design of High Affinity ... [https://www.biolscigroup.us/articles/GJBBS-3-109.php]
- 45. Biosensing Applications of Molecularly Imprinted-Polymer- ... [https://www.mdpi.com/2227-9717/12/1/177]
- 46. Recent advances in molecularly imprinted polymers toward ... [https://www.sciencedirect.com/science/article/abs/pii/S0956566325005111]
- 47. Development of Optimal Conditions for Synthesis ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12115306/]
- 48. Selective recognition of paracetamol using optimized deep ... [https://link.springer.com/article/10.1007/s10965-025-04547-1]
- 49. Synthesis and application of a molecularly imprinted ... [https://link.springer.com/article/10.1007/s10965-025-04458-1]
- 50. Molecular Imprinting on Nanozymes for Sensing Applications [https://pmc.ncbi.nlm.nih.gov/articles/PMC8152260/]
- 51. Molecularly Imprinted Polymers for the Selective ... [https://link.springer.com/article/10.1007/s42250-023-00740-1]
- 52. A Comprehensive Review of Nanozyme Construction ... [https://www.sciencedirect.com/science/article/pii/S2666053925001122]
- 53. A tutorial on the synthesis and applications of molecularly ... [https://www.sciencedirect.com/science/article/pii/S2772391725000428]
- 54. To Remove or Not to Remove? The Challenge of ... [https://www.mdpi.com/1422-0067/12/7/4327]
- 55. Characterization of the heterogeneous binding site affinity ... [https://www.sciencedirect.com/science/article/pii/S1570023204001321]
- 56. Strategies to remove templates from molecularly imprinted ... [https://www.sciencedirect.com/science/article/abs/pii/S0165993623005241]
- 57. Template-assisted synthesis of molecularly imprinted ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10173658/]
- 58. Molecularly imprinted polymers (MIPs): emerging biomaterials ... [https://biomaterialsres.biomedcentral.com/articles/10.1186/s40824-023-00388-5]
- 59. Assessing the In Vivo Biocompatibility of Molecularly Imprinted ... [https://pubmed.ncbi.nlm.nih.gov/36365575/]
- 60. Enhancing in vivo cell and tissue targeting by modulation ... [https://www.nature.com/articles/s41467-024-48442-7]
- 61. Sensing by Molecularly Imprinted Polymer: Evaluation of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6470915/]
- 62. MIPs As Chromatographic Stationary Phases for Molecular ... [https://sites.science.oregonstate.edu/chemistry/remcho/publications/analchem71.htm]
- 63. Scatchard plot: Common misinterpretation of binding ... [https://www.sciencedirect.com/science/article/abs/pii/0003269780901608]
- 64. Comparative study of molecularly imprinted polymer ... [https://www.sciencedirect.com/science/article/abs/pii/S0308814624001031]
- 65. Novel molecularly imprinted aerogels [https://www.sciencedirect.com/science/article/abs/pii/S0039914023007348]
- 66. A Novel Metal-Organic Framework Composite, MIL-101(Cr) ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6582831/]
- 67. Surface molecularly imprinted polymer based on core-shell ... [https://www.sciencedirect.com/science/article/abs/pii/S000326702030725X]
- 68. Development of Molecularly Imprinted Polymers via Dual ... [https://www.sciencedirect.com/science/article/pii/S266683192500181X]
- 69. Magnetic beta cyclodextrin molecularly imprinted polymer for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12432462/]
- 70. Surface imprinting polymerization for synthesis of a ... [https://link.springer.com/article/10.1007/s11696-025-04365-1]
- 71. Advances in biomass-based molecularly imprinted polymers [https://pubs.rsc.org/en/content/articlehtml/2025/gc/d5gc01018g]
- 72. Exploring molecularly imprinted polymers as artificial ... [https://www.sciencedirect.com/science/article/pii/S2773207X2200001X]
- 73. Molecularly imprinted polymers for pharmaceutical and ... [https://www.sciencedirect.com/science/article/pii/S2949771X24000148]