Strategies to Reduce Non-Specific Adsorption in Complex Serum Samples: A Guide for Biomedical Researchers
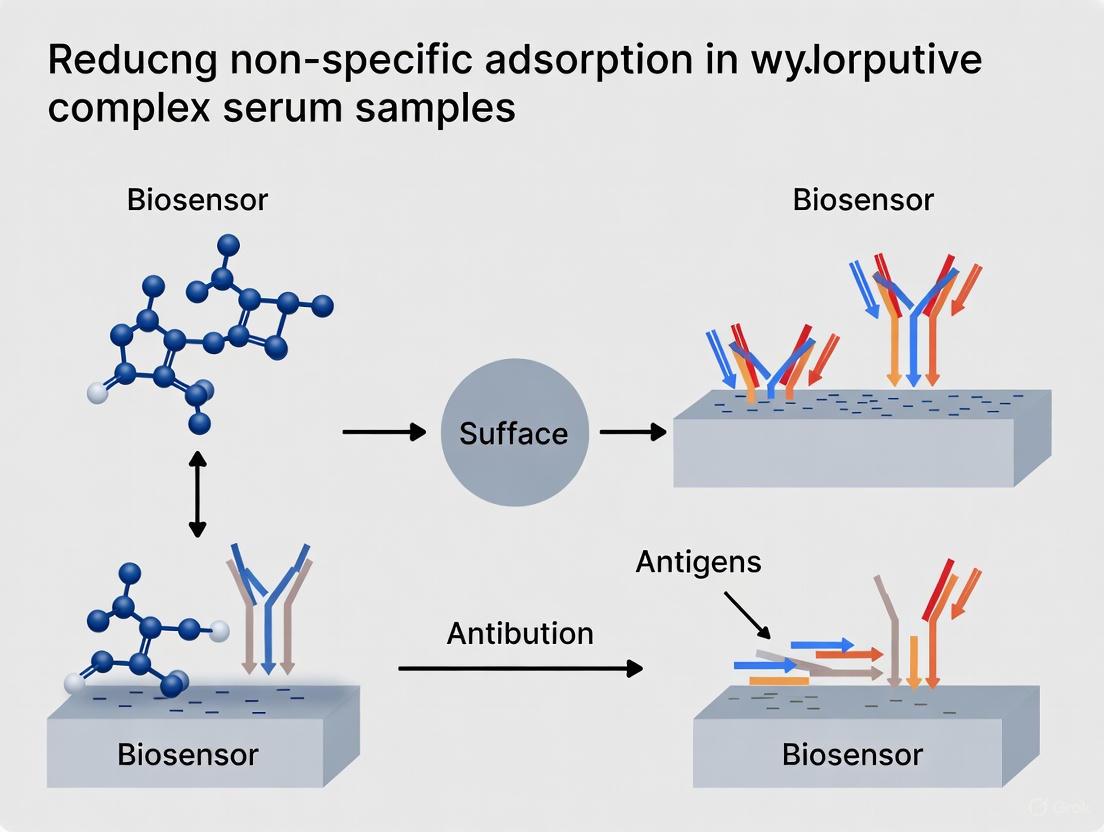
Non-specific adsorption (NSA) is a critical challenge that compromises the sensitivity, specificity, and reliability of biosensors and immunoassays used in complex biological matrices like serum.
Strategies to Reduce Non-Specific Adsorption in Complex Serum Samples: A Guide for Biomedical Researchers
Abstract
Non-specific adsorption (NSA) is a critical challenge that compromises the sensitivity, specificity, and reliability of biosensors and immunoassays used in complex biological matrices like serum. This article provides a comprehensive guide for researchers and drug development professionals, covering the foundational mechanisms of NSA, practical methodologies for its suppression, advanced troubleshooting protocols, and rigorous validation techniques. By synthesizing current research on antifouling coatings, surface chemistry, and sample pre-treatment, this resource aims to equip scientists with the knowledge to design robust assays and biosensors for accurate analyte detection in serum, ultimately enhancing the translation of diagnostic and research tools from the lab to the clinic.
Understanding the Enemy: The Fundamentals of Non-Specific Adsorption in Serum
Defining Non-Specific Adsorption and Its Impact on Assay Performance
Non-specific adsorption (NSA) is a pervasive challenge in bioanalytical science that compromises the accuracy, sensitivity, and reliability of experimental results. For researchers working with complex serum samples, NSA presents a significant barrier to obtaining clean data and reproducible assays. This technical resource center provides practical guidance to help scientists identify, troubleshoot, and mitigate NSA in their experimental workflows, with particular emphasis on applications in drug development and clinical research.
FAQs: Understanding the Fundamentals
What is non-specific adsorption and how does it affect my assays?
Non-specific adsorption refers to the undesirable adhesion of atoms, ions, or molecules to surfaces through non-covalent bonding forces rather than specific biorecognition events [1]. Unlike specific binding, NSA occurs through physisorption (physical adsorption) driven by:
In biosensing applications, NSA causes elevated background signals that are indistinguishable from specific binding, leading to:
- False-positive results and compromised specificity [1]
- Reduced sensitivity and impaired detection limits [1] [2]
- Decreased dynamic range and poor reproducibility [1]
- Sensor drift and signal degradation over time [2]
Why are serum samples particularly problematic for NSA?
Serum presents exceptional challenges due to its complex composition, containing 40-80 mg/mL of proteins alongside lipids, metabolites, and other biomolecules [3]. The high concentration of diverse proteins creates intense competition for surface binding sites, while the varied physicochemical properties of serum components enable multiple adsorption mechanisms to occur simultaneously [2] [4].
What types of analytes are most susceptible to NSA?
Certain classes of analytes demonstrate particularly high susceptibility to NSA:
| Analyte Type | Key Characteristics Promoting NSA | Common Applications |
|---|---|---|
| Phosphorylated Compounds | Acidic phosphate groups interact with metal surfaces [5] | Metabolic studies, kinase assays |
| Nucleic Acids/Oligonucleotides | Phosphate backbone, amphoteric nature [5] [4] | Genetic testing, therapeutic oligonucleotides |
| Peptides/Proteins | Amphoteric amino acids, charged groups, hydrophobic regions [4] | Biomarker detection, immunoassays |
| Cationic Lipids | Positively charged head groups, hydrophobic tails [4] | Drug delivery systems, lipidomics |
| Small Molecules with Acidic Groups | Carboxylate, phosphate, or other acidic moieties [5] | Pharmaceutical compounds, metabolites |
Troubleshooting Guides
Diagnosing NSA in Experimental Results
Recognizing the signature patterns of NSA is the first step toward resolution:
| Symptom | Common Manifestations | Recommended Investigations |
|---|---|---|
| Poor Recovery | Inconsistent extraction recovery calculations; higher signal at high concentrations and lower at low concentrations [4] | Compare results across concentration range; use low-adsorption materials |
| Signal Anomalies | Elevated background, signal drift, system carryover [4] | Include appropriate controls; analyze blank samples |
| Chromatographic Issues | Peak tailing, loss of intensity, poor peak shape [5] [4] | Use low-adsorption columns; modify mobile phase |
| Inconsistent Results | Poor reproducibility between replicates or experiments [1] [5] | Standardize sample handling; implement surface passivation |
Systematic Workflow for NSA Investigation
The following diagram outlines a logical approach to identifying and addressing NSA problems:
Effective Mitigation Strategies for Serum Samples
Surface Passivation and Blocking Methods
Protein-Based Blockers:
- BSA (Bovine Serum Albumin): Traditional blocking agent that occupies vacant surface sites [1]
- Casein and Milk Proteins: Effective for ELISA, Western blotting, and enzyme-based assays [1]
- Serum Incubation: Pre-adsorption with diluted serum from same species as sample
Synthetic Surface Chemistries:
- Polyethylene Glycol (PEG): Creates hydrated barrier that sterically hinders approach of biomolecules [1] [3]
- Zwitterionic Peptide SAMs: Afficoat and similar technologies use alternating charged groups for ultra-low fouling surfaces [3]
- Dextran Hydrogels: Provides three-dimensional hydrophilic matrix that resists protein adsorption [6]
- Surface-Initiated Polymerization (SIP): Demonstrated superior performance with minimal NSA in comparative studies [6]
Mobile Phase and Buffer Modifications
Surfactant Additives:
- Anionic Surfactants: Sodium dodecyl sulfate (SDS) effectively eliminates NSA in molecularly imprinted polymers [7]
- Cationic Surfactants: Cetyl trimethyl ammonium bromide (CTAB) modifies surface charge to reduce unwanted interactions [7]
- Non-ionic Surfactants: Tween and Triton series provide milder detergent effects with less method interference [4]
Competitive Binding Agents:
- Metal Chelators: EDTA reduces adsorption of phosphorylated and nucleic acid compounds by sequestering metal ions [4]
- Carrier Proteins: Addition of BSA or other proteins to samples and standards competes for binding sites [4]
- Ionic Additives: Increased salt concentration can shield electrostatic interactions
Hardware and Consumable Selection
Low-Adsorption Materials:
- PEEK (Polyether Ether Ketone): Alternative to stainless steel in LC systems, especially for acidic compounds [5]
- Titanium Components: Reduced metal interaction compared to traditional stainless steel [5]
- Hybrid Surface Technology: MaxPeak HPS and similar technologies create barrier layers that prevent contact with metal surfaces [5] [8]
- Surface-Passivated Consumables: Low-adsorption tubes and plates specifically designed for proteins and nucleic acids [4]
Experimental Protocols for NSA Evaluation
Protocol 1: Quantitative NSA Assessment in Serum-Containing Samples
Materials:
- Complex biological sample (serum, cell lysate, etc.)
- Appropriate biosensor platform (SPR, QCM, etc.) or analytical system
- Reference surface (blocked/passivated) and test surface
- Buffer systems for dilution and washing
Procedure:
- Prepare serial dilutions of serum in relevant buffer (typically 1:10 to 1:100)
- Establish baseline signal with buffer alone
- Expose surface to serum dilution for predetermined time (typically 15-30 minutes)
- Rinse with buffer and measure residual signal
- Quantify adsorbed mass using appropriate calibration standards
- Compare results across different surface chemistries
Interpretation: Surfaces demonstrating <5% signal increase over baseline are considered excellent, while >15% indicates significant NSA problems [6] [3].
Protocol 2: Surface Passivation with Zwitterionic SAMs
Materials:
- Gold sensor surfaces or other appropriate substrates
- Afficoat solution or alternative zwitterionic thiol compounds
- Ethanol for cleaning
- Peptide coupling reagents (EDC/NHS) if subsequent functionalization required
Procedure:
- Thoroughly clean gold surfaces with ethanol and dry under nitrogen
- Incubate with Afficoat solution (typically 0.1-1.0 mM in ethanol) for 12-24 hours
- Rinse extensively with ethanol and water to remove unbound thiols
- Characterize surface with FTIR or contact angle measurement
- Test NSA resistance with serum samples as described in Protocol 1
Performance Expectations: Properly prepared Afficoat surfaces demonstrate >80% reduction in NSA compared to unmodified gold and outperform traditional PEG coatings [3].
Research Reagent Solutions
| Reagent/Category | Specific Examples | Mechanism of Action | Applicable Sample Types |
|---|---|---|---|
| Blocking Proteins | BSA, Casein, Milk Proteins | Occupies vacant surface sites through preferential adsorption | Serum, plasma, cell culture media [1] |
| Polymer Coatings | PEG, Dextran, PVPA | Creates hydrated physical barrier that resists protein approach | Complex biological fluids [1] [6] |
| Zwitterionic SAMs | Afficoat, Peptide SAMs | Presents alternating charged groups for ultra-low fouling | Crude cell lysate, serum, plasma [3] |
| Surfactants | SDS, CTAB, Tween-20 | Modifies surface charge and disrupts hydrophobic interactions | Urine, bile, CSF [7] [4] |
| Chelating Agents | EDTA, Citrate, Phosphate | Sequesters metal ions to prevent metal-mediated adsorption | Phosphorylated compounds, nucleic acids [5] [4] |
| Low-Adsorption Materials | PEEK, Titanium, Hybrid Surfaces | Reduces available interaction sites through surface passivation | All sample types, especially problematic analytes [5] [8] |
Advanced Applications in Biosensing
SPR Biosensor Applications
Surface Plasmon Resonance (SPR) biosensors are particularly vulnerable to NSA effects while also offering powerful capabilities for real-time interaction monitoring [9]. Successful implementation with serum samples requires:
Surface Chemistry Optimization:
- CM-Dextran: Traditional SPR surface with carboxylated groups for biomolecule immobilization
- PEG-Based Layers: Reduced fouling compared to dextran [3]
- Zwitterionic Technologies: Afficoat demonstrates superior performance with 3-5× lower NSA compared to alternatives [3]
Regeneration Protocols:
- High-Salt Washes: Disrupts electrostatic interactions (e.g., 1-2 M NaCl)
- Mild Detergents: Removes hydrophobically-bound contaminants (e.g., 0.05% Tween-20)
- Acidic/Basic Eluents: Regenerates surfaces through pH manipulation (e.g., 10 mM glycine pH 2.5)
Method Validation and Quality Control
Establishing robust NSA monitoring protocols ensures long-term assay reliability:
Positive Controls:
- Include known problematic compounds to verify surface performance
- Monitor baseline drift during extended runs
- Track carryover between samples
Performance Metrics:
- LOD/LOQ shifts: Indicator of rising background interference
- Standard curve linearity: Degradation suggests progressive surface fouling
- Recovery consistency: Inconsistent extraction efficiency signals adsorption issues
Successfully managing non-specific adsorption in serum samples requires a systematic approach that addresses all three fundamental factors: the solid surfaces, solution composition, and analyte properties. By implementing the diagnostic strategies, mitigation approaches, and validation protocols outlined in this technical guide, researchers can significantly improve data quality and assay reproducibility in even the most challenging biological matrices.
Key Interfering Components in Human Serum and Plasma
Frequently Asked Questions
What are the most common causes of non-specific adsorption (NSA) in serum and plasma samples? NSA is primarily caused by physisorption of biomolecules to surfaces through hydrophobic forces, ionic interactions, van der Waals forces, and hydrogen bonding [1]. Common interferents include proteins like human gamma globulin, complement proteins, and lipids [10]. The presence of autoantibodies (e.g., rheumatoid factor) and human anti-animal antibodies (HAMA) can also lead to significant assay interference [10].
My immunoassay shows high background signal. What could be the cause? High background signal often results from non-specific adsorption of serum components to the assay surface or components [1]. This can be due to matrix effects from sample components like bilirubin, hemoglobin, or cholesterol [10]. Other causes include cross-reactivity with structurally similar molecules, heterophilic antibodies, or insufficient blocking of the assay surface [10].
How can I minimize non-specific binding in my biosensor assays? Using surface coatings like polyethylene glycol (PEG), dextran, or surface-initiated polymerization can create a hydrophilic, non-fouling boundary layer that reduces NSA [6] [1]. Incorporating blocking agents such as bovine serum albumin (BSA), casein, or normal serum can also help saturate potential interfering sites [10] [1].
Why do I get different results between one-stage and two-stage factor activity assays? These assays differ in methodology and susceptibility to interference. One-stage clotting assays can be affected by pre-activation of factors or the presence of antiphospholipid antibodies like lupus anticoagulant [11]. Chromogenic (two-stage) assays avoid some limitations of one-stage assays by using a different detection system that is less prone to certain interferences [12].
Troubleshooting Guide
Problem: High Background Signal in Immunoassays
| Possible Cause | Diagnostic Tests | Solutions |
|---|---|---|
| Matrix Effects | Spike and recovery experiments [10] | Dilute sample; use matrix-matched standards; modify assay buffer pH/ionic strength [10] |
| Heterophilic Antibodies | Test with heterophilic antibody blocking reagents [10] | Add blocking agents (normal serum, HAMA blockers) [10] |
| Insufficient Blocking | Compare background with different blocking agents | Use protein blockers (BSA, casein, milk proteins) [10] [1] |
| Cross-reactivity | Test analyte specificity with related compounds | Use more specific antibodies; change assay format [10] |
Problem: Inconsistent Results Between Assay Platforms
| Possible Cause | Diagnostic Tests | Solutions |
|---|---|---|
| Drug Interference | Review patient medication history | Use alternative assay formats; consult literature for known drug interactions [10] |
| Biotin Interference | Check for biotin supplement use | Use biotin-free assays; ask patients to pause supplements [10] |
| Sample Preparation Issues | Compare fresh vs. stored samples | Standardize sample collection; avoid repeated freeze-thaw cycles [5] |
| Hook Effect | Test sample at multiple dilutions | Dilute sample and re-assay [10] |
Key Interfering Components and Mitigation Strategies
The table below summarizes major interfering components found in human serum and plasma and recommended mitigation approaches.
| Interfering Component | Source/Description | Impact on Assays | Mitigation Strategies |
|---|---|---|---|
| Human Anti-Animal Antibodies (HAAA) | Human antibodies against animal immunoglobulins [10] | False positives/negatives by binding assay antibodies [10] | Heterophilic antibody blockers; species-specific serum [10] |
| Rheumatoid Factor | Autoantibody targeting IgG Fc portion [10] | Binds to assay immunoglobulins, causing unreliable signals [10] | Use RF-specific blocking reagents; Fab fragments [10] |
| Biotin | High-dose supplements [10] | Interferes in streptavidin-biotin detection systems [10] | Pause supplements; use biotin-free assays [10] |
| Complement Proteins | Serum proteins in innate immune system [10] | Non-specific binding to assay components [10] | Use complement-inactivated serum; EDTA plasma [10] |
| Lipids (Lipaemia) | High triglyceride levels [10] | Light scattering; non-specific binding [10] | Sample dilution; ultracentrifugation; use of clearing agents [10] |
| Hemoglobin | Hemolysis of blood samples [10] | Color interference; peroxidase activity in ELISA [10] | Avoid hemolyzed samples; use proper sample handling [10] |
| Bilirubin | Liver dysfunction; hemolysis [10] | Color interference in colorimetric assays [10] | Sample dilution; blank correction; use of antioxidant [10] |
Experimental Protocols
Protocol 1: Spike and Recovery Experiment for Interference Testing
Purpose: To assess whether components in a sample matrix interfere with accurate analyte detection [10].
Materials Needed:
- Test sample matrix (serum/plasma)
- Pure analyte standard
- Assay buffer
- Appropriate immunoassay reagents
Procedure:
- Prepare three sets of samples in duplicate or triplicate:
- Neat matrix: Sample matrix with no spike (determines endogenous analyte levels)
- Spiked buffer (control): Known concentration of analyte spiked into assay buffer
- Spiked matrix (test): Same concentration of analyte spiked into sample matrix
Run all samples according to assay protocol.
Calculate percentage recovery: % Recovery = (Concentration in Spiked Matrix / Concentration in Spiked Buffer) × 100
Interpret results:
- 80-120% recovery: Acceptable, minimal interference
- <80% recovery: Signal suppression (matrix interference)
- >120% recovery: Signal enhancement (possible interference or cross-reactivity) [10]
Protocol 2: Assessment of Non-Specific Adsorption on Biosensor Surfaces
Purpose: To compare non-specific adsorption of serum and cell lysate on different biosensor surfaces [6].
Materials Needed:
- SPRi (Surface Plasmon Resonance Imaging) biosensor
- Different surface chemistries (PEG, cyclodextrin, dextran, SIP)
- Human serum and cell lysate samples
- MALDI-TOF/TOF MS for surface evaluation [6]
Procedure:
- Fabricate biosensor surfaces with different chemistries (PEG, α-cyclodextrin, hydrogel dextran, SIP-based gold surfaces).
- Confirm surface fabrication using FTIR spectroscopy [6].
- Apply human serum and cell lysate samples to different surfaces.
- Use SPRi to measure non-specific adsorption response.
- Evaluate surfaces with MALDI-TOF/TOF MS technique [6].
- Compare results across surfaces - SIP and dextran surfaces typically show minimum non-specific adsorption [6].
Research Reagent Solutions
| Reagent Category | Specific Examples | Function/Application |
|---|---|---|
| Blocking Agents | BSA, Casein, Normal Serum (various species) [10] | Reduce NSA by saturating potential interfering sites on surfaces [10] [1] |
| Heterophilic Antibody Blockers | HAMA Blocking Reagent, Species-Specific Sera [10] | Reduce interference from human anti-animal antibodies [10] |
| Matrix Effect Controls | Conjugated/Unconjugated Bilirubin, Haemoglobin, Cholesterol [10] | Identify and quantify specific matrix interference [10] |
| Reference Materials | Normal Human Serum (various ages), Rheumatoid Factor Control [10] | Provide standardized controls for assay validation [10] |
| Surface Coatings | PEG, Dextran, Surface Initiated Polymerization [6] | Create non-fouling surfaces to minimize NSA in biosensors [6] |
Experimental Workflow and Interference Mechanisms
Interference Troubleshooting Workflow
Serum Interference Mechanisms
Physical and Chemical Mechanisms Driving NSA
Frequently Asked Questions (FAQs)
FAQ 1: What is non-specific adsorption (NSA) and how does it affect my biosensor's performance? Non-specific adsorption (NSA) refers to the unwanted accumulation of molecules (e.g., proteins, lipids) from your sample onto the biosensor's surface. This is distinct from the specific binding of your target analyte to its bioreceptor. In complex samples like serum, NSA can severely impact your results by [2]:
- Causing false positives: Non-specifically adsorbed molecules generate a background signal that is indistinguishable from your specific analyte signal.
- Causing false negatives: Fouling can block access to the bioreceptor or restrict its ability to change conformation, preventing target binding.
- Reducing sensitivity and reproducibility: NSA degrades the sensor surface over time, leading to signal drift and unreliable data.
FAQ 2: What are the primary physical and chemical forces responsible for NSA? NSA is primarily driven by physisorption (physical adsorption), which involves a combination of weak intermolecular forces between the sensor surface and components in the sample matrix. The main mechanisms are [2] [1]:
- Electrostatic interactions between charged surfaces and charged protein residues.
- Hydrophobic interactions between non-polar surface areas and hydrophobic protein domains.
- Hydrogen bonding or other dipole-dipole interactions.
- van der Waals forces.
FAQ 3: My research involves human serum samples. Why is this matrix particularly challenging? Serum is a complex biological fluid containing a high concentration of diverse proteins, with human serum albumin (HSA) being the most abundant. These proteins readily adsorb to surfaces through the mechanisms described above. Furthermore, serum contains other interfering components like lipids and salts. A specific challenge is that inflammatory conditions can elevate proteins like C-reactive protein (CRP), which can form oxidative cross-links with HSA, creating stable, fouling complexes on your sensor [13].
FAQ 4: Are there ways to actively remove adsorbed molecules after fouling occurs? Yes, alongside passive blocking methods, active removal methods are an area of development. These methods generate forces to shear away weakly adsorbed biomolecules [1]. They can be categorized as:
- Transducer-based: Using electromechanical or acoustic energy to create surface waves that displace foulants.
- Fluid-based: Leveraging hydrodynamic flow within microfluidic channels to create shear forces that wash away non-specifically bound molecules.
Troubleshooting Guides
Problem: High Background Signal in Serum Samples
A high and variable background signal is one of the most common symptoms of NSA. The following workflow helps diagnose and address this issue.
Guide: Selecting Materials to Minimize NSA
The material of your sensor surface or microfluidic channel is a critical first line of defense. The intrinsic properties of the material, such as hydrophilicity and terminal functional groups, greatly influence protein adsorption. The table below summarizes experimental data on the non-specific adsorption of Bovine Serum Albumin (BSA) to various materials, providing a quantitative comparison for your selection process [14].
| Material | Surface Characteristics | Relative BSA Adsorption (Fluorescence Intensity) | Key Rationale |
|---|---|---|---|
| SU-8 | Hydrophilic (after cleaning) | ~50 (Lowest) | Hydrophilicity reduces protein adsorption. |
| CYTOP S-grade | Terminal -CF₃ group | ~120 | Low surface energy and terminal trifluoromethyl group. |
| CYTOP M-grade | Terminal amide-silane group | ~190 | Higher adsorption compared to S-grade. |
| CYTOP A-grade | Terminal carboxyl group | ~210 | Charged functional groups can increase interaction. |
| Silica (SiO₂) | Hydrophilic but with fixed positive charge | ~160 (Unexpectedly High) | Fixed positive charge attracts negatively charged BSA. |
Guide: Implementing Antifouling Surface Coatings
Passive methods involve coating the surface with a physical or chemical layer that prevents foulants from adsorbing. The goal is to create a thin, hydrophilic, and neutrally charged boundary layer [2] [1].
Detailed Protocol: Blocking with Bovine Serum Albumin (BSA)
- Principle: BSA molecules adsorb to unfunctionalized and "sticky" sites on the surface, effectively blocking them from subsequent adsorption of interfering proteins in your sample [1].
- Materials:
- Bovine Serum Albumin (BSA), fraction V.
- Phosphate Buffered Saline (PBS), pH 7.4.
- Incubation chamber (e.g., flow cell, multi-well plate).
- Washing buffer (e.g., PBS with 0.05% Tween 20).
- Procedure:
- After immobilizing your bioreceptor (e.g., antibody) and washing the surface, prepare a blocking solution of 1-5% (w/v) BSA in PBS.
- Completely immerse or flow the blocking solution over the sensor surface.
- Incubate for 30-60 minutes at room temperature.
- Thoroughly wash the surface with washing buffer to remove unbound BSA.
- The sensor is now ready for use with your sample. Note that BSA blocking is typically irreversible.
Detailed Protocol: Reversible Blocking with Amphiphilic Sugars
- Principle: Amphiphilic molecules like n-Dodecyl β-D-maltoside competitively and reversibly adsorb to hydrophobic surfaces. Their hydrophilic sugar headgroups create a barrier to NSA, and they can be washed away without permanent surface modification [15].
- Materials:
- n-Dodecyl β-D-maltoside (DDM).
- Your standard assay buffer (e.g., PBS).
- The analyte sample.
- Procedure:
- Prepare your analyte sample and standards as usual.
- Add DDM to the sample solution directly to a final concentration above its critical micelle concentration (CMC). (Note: The exact concentration must be optimized for your system).
- Perform the assay as normal. The DDM in the solution will dynamically block free surfaces during the measurement.
- After the assay, a thorough wash will remove the DDM, returning the surface to its original state for the next experiment.
The Scientist's Toolkit: Key Research Reagent Solutions
| Reagent / Material | Function in NSA Reduction | Key Consideration |
|---|---|---|
| Bovine Serum Albumin (BSA) | Protein-based blocking agent; physically adsorbs to vacant sites [1]. | Standard, low-cost method; can be difficult to remove (irreversible). |
| Casein / Milk Proteins | Protein-based blocker; effective for ELISA and Western blotting [1]. | Similar to BSA; ensure compatibility with your detection system. |
| n-Dodecyl β-D-maltoside | Amphiphilic sugar; reversible surface blocker when added to solution [15]. | Enables simple surface chemistry; requires optimization of concentration. |
| SU-8 Epoxy Resist | Hydrophilic polymer for microfluidics; exhibits low intrinsic NSA [14]. | Ideal for fabricating microfluidic channels; requires UV lithography. |
| CYTOP S-grade | Fluoropolymer with -CF₃ terminal group; low refractive index and low NSA [14]. | Excellent for optical biosensors; low adhesion may require surface activation. |
| Specific Peptides / Zwitterionic Polymers | Engineered antifouling coatings; form highly hydrated, neutral surfaces [2]. | High-performance modern materials; may require complex synthesis/fabrication. |
Troubleshooting Guides
Guide 1: Addressing Non-Specific Adsorption (NSA) in Biosensor Analysis of Serum Samples
Problem Statement: High background signal, false positives, or reduced sensitivity when analyzing complex serum samples with a biosensor. The signal is unstable or drifts over time, compromising data accuracy [1] [2].
Core Issue: NSA, or biofouling, occurs when proteins and other biomolecules from serum physisorb onto your sensing interface. This fouling layer can block analyte access, interfere with electron transfer (in EC biosensors), or create a signal indistinguishable from specific binding (in SPR biosensors) [1] [2]. The mechanisms driving this include hydrophobic interactions, electrostatic forces, hydrogen bonding, and van der Waals forces [2].
Troubleshooting Steps:
Evaluate and Optimize Your Antifouling Coating:
- Action: If you are using a self-assembled monolayer (SAM) or polymer coating like PEG, ensure it forms a dense, hydrophilic, and neutrally charged barrier. Inadequate coating is a primary failure point [1].
- Verification: Characterize your coated surface using techniques like FTIR or SPRi to confirm successful modification before proceeding with assays [6].
Implement a Robust Blocking Step:
- Action: After immobilizing your bioreceptor, incubate the sensor with a blocking solution. Common blockers for serum samples include serum albumin (e.g., BSA), casein, or specialized commercial protein-free blocking reagents [1] [16].
- Protocol: Incubate for 1 hour at room temperature with a gentle agitation. Follow the manufacturer's recommendations if using a commercial blocker [16].
Optimize Your Sample and Running Buffer:
- Action: Supplement your assay buffer with additives that reduce NSA. This can include a small percentage of a detergent (e.g., Tween 20) or a commercial sample diluent specifically formulated to reduce matrix interferences [16] [2].
- Protocol: For initial optimization, try a buffer containing 0.05% Tween 20 and 1% BSA. Centrifuge serum samples before analysis to remove particulates [2].
Employ Active Removal Methods (if applicable to your system):
- Action: In microfluidic systems, consider applying active NSA removal techniques. These generate shear forces at the sensor surface to physically dislodge weakly adsorbed molecules [1].
- Protocol: Hydrodynamic methods use controlled fluid flow, while transducer-based methods use electromechanical or acoustic energy. These typically require specialized instrument setups [1].
Prevention Checklist: ☐ All surfaces (sensing and fluidic) are properly coated with an antifouling material. ☐ A effective blocking step is included in the assay protocol. ☐ Samples are centrifuged and prepared in a compatible, optimized buffer. ☐ Washing steps are sufficient but not overly aggressive [17].
Guide 2: Minimizing False Positives in ADP Detection Assays for High-Throughput Screening (HTS)
Problem Statement: Artificially inflated hit rates in kinase, ATPase, or other ATP-dependent enzyme screens due to compounds that interfere with the assay's detection system rather than genuinely inhibiting the target enzyme [18].
Core Issue: In indirect or coupled assays (e.g., those using luciferase to detect ATP/ADP conversion), test compounds can inhibit the coupling enzymes or directly interfere with the optical signal (e.g., by quenching luminescence or autofluorescence), leading to false-positive inhibition readouts [18] [19].
Troubleshooting Steps:
Switch to a Direct Detection Assay Format:
- Action: The most effective solution is to replace a coupled assay with a method that directly detects the primary product, ADP. This eliminates the extra layers where interference can occur [18].
- Protocol: Implement a homogeneous, "mix-and-read" immunoassay that uses a fluorescent tracer and an antibody against ADP. The signal is generated by competitive displacement of the tracer by ADP, directly correlating with enzyme activity [18].
If a Coupled Assay is Necessary, Run a Counterassay:
- Action: To identify false positives, run a counterassay that contains all detection reagents but lacks the primary target enzyme. Any compound that shows a signal in this counterassay is an interferent [18] [19].
- Protocol: In a separate plate well, mix the test compound with the luciferase/enzyme coupling system. A change in signal indicates direct compound interference with the detection system [19].
Red-Shift Your Detection Wavelength:
- Action: If using fluorescence, design your assay with red-shifted readouts (>500 nm). Compound libraries have a much lower frequency of autofluorescent molecules in the red region compared to the blue/green spectrum [19].
- Protocol: Choose assay kits that use fluorophores with excitation/emission in the far-red (e.g., Cy5, Alexa Fluor 647) to minimize background from compound autofluorescence [19].
Perform a Pre-Read in Kinetic Assays:
- Action: Before initiating the enzymatic reaction, take a fluorescence pre-read of the plate with all components, including test compounds.
- Protocol: This initial read measures the intrinsic fluorescence of the compound library. You can then flag or filter out compounds with high initial fluorescence before analyzing the assay results [19].
Comparison of ADP Detection Methods
| Assay Type | Detection Mechanism | Key Advantage | Key Disadvantage | False Positive Risk |
|---|---|---|---|---|
| Coupled Enzyme (Luminescent) | Multiple enzymes convert ADP to ATP, driving a luciferase reaction [18]. | Highly sensitive, widely adopted [18]. | Multiple points for compound interference (e.g., luciferase inhibition) [18]. | High [18] |
| Colorimetric (Malachite Green) | Detects inorganic phosphate (Pi) released from ATP [18]. | Low cost, simple setup [18]. | Interference from colored compounds and phosphate buffers; low sensitivity [18]. | Moderate [18] |
| Direct Fluorescent Immunoassay | Fluorescent tracer is displaced from an anti-ADP antibody by ADP produced in the reaction [18]. | Homogeneous, "mix-and-read"; minimal interference points [18]. | Requires optimization of tracer/antibody concentration [18]. | Very Low [18] |
Frequently Asked Questions (FAQs)
What are the primary causes of non-specific adsorption (NSA) in biosensors?
NSA is primarily caused by physisorption, where molecules from your sample (like serum proteins) adhere to the sensor surface through a combination of hydrophobic interactions, electrostatic forces, hydrogen bonding, and van der Waals forces [1] [2]. This is distinct from the specific, covalent-like binding (chemisorption) you design for your bioreceptors.
What is the difference between passive and active methods for reducing NSA?
- Passive Methods aim to prevent adsorption by creating a physical or chemical barrier on the sensor surface. This includes coating the surface with blocker proteins (e.g., BSA), polymers (e.g., PEG), or hydrogel layers (e.g., dextran) that repel biomolecules [1].
- Active Methods dynamically remove adsorbed molecules after they have attached to the surface. This is typically done by generating surface shear forces, either through fluid flow (hydrodynamic) or with transducers (acoustic or electromechanical), to overpower the adhesive forces of the foulants [1].
Our ELISA for serum biomarkers has a high background. What are the first three things I should check?
- Blocking: Ensure you have an effective and complete block step. If using BSA, try switching to a commercial, protein-free blocking reagent for potentially better performance [16].
- Washing: Confirm your wash buffer is fresh and that you are performing an adequate number of wash steps. However, avoid overly aggressive washing that could displace detection antibodies [16] [17].
- Sample Diluent: Use a specialized sample/assay diluent designed to reduce matrix interferences from serum, rather than a standard buffer. These diluents can significantly cut down cross-reactivity and false positives [16].
A large percentage of our HTS hits from a fluorescent assay were fluorescent compounds. How can we prevent this?
This is a common issue. Your strategy should include:
- Assay Redesign: For future screens, use assays with red-shifted fluorescent readouts (emission >500 nm), as chemical libraries have far fewer fluorescent compounds in this region [19].
- Hit Triage: Implement a counterassay that detects the fluorescent signal in the absence of the biological target. This will immediately flag autofluorescent compounds and quenchers [19].
- Orthogonal Validation: Always confirm hits from a primary fluorescent screen using an assay with a different detection technology (e.g., AlphaScreen, SPR, or a direct biochemical assay) [18] [19].
What are some key reagent solutions for improving assay specificity and reducing false positives?
Research Reagent Solutions for Complex Samples
| Reagent / Material | Primary Function | Example Use Case |
|---|---|---|
| PEG-based Coatings | Forms a hydrated, neutral polymer brush that sterically repels proteins [1] [6]. | Creating non-fouling surfaces on biosensors (SPR, electrochemical) and microfluidic channels [6]. |
| Commercial Blocking Reagents | Adsorbs to surface vacancies, shielding them from non-specific protein binding [16]. | Reducing background in ELISA and immunosensor assays after antibody immobilization [16]. |
| Specialized Sample Diluents | Contains agents that reduce matrix effects, mask interfering factors, and minimize NSA [16]. | Diluting complex samples like serum or cell lysate for analysis in immunoassays [16]. |
| Surface-Initiated Polymerization | Grows a dense, highly controllable polymer film on the sensor surface for superior antifouling properties [6]. | Advanced biosensor platforms (e.g., SPRi) requiring extreme resistance to fouling from serum and cell lysate [6]. |
| Direct Immunoassay Kits | Detects the direct product of a reaction (e.g., ADP) via immunodetection, avoiding multi-enzyme coupling [18]. | High-throughput screening of kinases/ATPases to minimize false positives from compound-interference with coupling enzymes [18]. |
Experimental Workflows & Visualizations
Workflow for Evaluating Antifouling Coatings
This diagram outlines a systematic protocol for developing and testing new antifouling surfaces for biosensors, particularly for use with complex samples like serum.
Mechanisms and Impact of Non-Specific Adsorption
This diagram illustrates how non-specifically adsorbed molecules interfere with different types of biosensor signals, leading to false positives and reduced sensitivity.
Practical Strategies for Suppressing Non-Specific Binding
In the analysis of complex serum samples for research and drug development, nonspecific adsorption (NSA) and sample complexity are significant barriers to obtaining accurate, reliable data. Nonspecific adsorption refers to the accumulation of species other than the target analyte on sensing interfaces, which can compromise signal stability, selectivity, and sensitivity [2]. Serum is a particularly challenging matrix because a small number of highly abundant proteins, such as albumin and immunoglobulins, can constitute the majority of the total protein content, masking lower-abundance proteins that are often critical biomarkers [20] [21]. Abundant protein depletion is therefore a vital pre-treatment step to reduce this complexity, minimize NSA, and enhance the detection of low-abundance analytes in applications from mass spectrometry to biosensing.
This technical support center provides troubleshooting guides and FAQs to help researchers navigate common issues encountered during abundant protein depletion protocols.
## Frequently Asked Questions (FAQs)
1. Why is abundant protein depletion necessary before analyzing serum samples?
Serum and plasma are dominated by a handful of highly abundant proteins, like albumin and IgG, which can account for over 50% of the total protein content [21]. This overwhelming abundance can obscure the detection of lower-concentration proteins (potential biomarkers) in analytical techniques like mass spectrometry. Depletion removes these top proteins, thereby reducing sample complexity and dynamic range, which allows for the enhanced identification and quantification of less abundant proteins [20] [22].
2. What is the typical efficiency of commercial depletion kits, and how is it measured?
Efficiency varies by product but can be very high. For example, some immunoaffinity-based spin columns are reported to remove >95% of albumin and IgG, and >99% of up to 14 abundant proteins [20]. Efficiency is typically confirmed using techniques like:
- Targeted Mass Spectrometry (MS): To quantify the removal of specific proteins.
- Enzyme-Linked Immunosorbent Assay (ELISA): To verify the depletion percentage of particular proteins like albumin [20].
- BCA Protein Assay: To estimate the total protein amount remaining in the depleted fraction (flow-through) [20].
3. My depletion protocol resulted in low recovery of my target protein. What could have gone wrong?
Low recovery can stem from several issues:
- Non-Specific Binding: Your target protein may be non-specifically adsorbing to the depletion resin or column hardware [7] [23].
- Overloading: Exceeding the sample volume capacity of the column can cause premature breakthrough of both abundant and target proteins [22].
- Insufficient Elution (for target recovery): If you are attempting to recover your target from the column, the elution conditions (e.g., buffer strength, pH, volume) may be inadequate [22].
- Carryover: If columns are reused, cross-contamination or carryover of abundant proteins from previous runs can occur [20].
4. How can I minimize non-specific adsorption of my target analyte during the depletion process?
Minimizing NSA is a multi-faceted challenge. Strategies include:
- Optimizing Buffer Composition: Introduce surfactants, salts, or carrier proteins into the binding and wash buffers to block non-specific sites on the resin and equipment surfaces [2].
- Using Passivated Materials: Employ low-protein-binding plastics and tubes throughout the procedure.
- Electrostatic Modification: In some specialized applications, modifying materials with charged surfactants (e.g., SDS, CTAB) has been shown to effectively eliminate non-specific adsorption by reacting with external functional groups [7].
## Troubleshooting Guide
The table below outlines common problems, their potential causes, and solutions.
| Problem | Potential Causes | Recommended Solutions |
|---|---|---|
| Low Depletion Efficiency | Column overloading; Incorrect buffer/pH; Expired or degraded resin | Do not exceed recommended sample volume; Verify buffer composition and pH; Use fresh reagents and columns [20] [22] |
| High Background in Analysis | Incomplete washing; Carryover from previous runs; Sample debris | Increase wash buffer volume/cycles; Use single-use columns or stringent regeneration; Centrifuge or filter sample pre-load [22] |
| Clogged Column / High Pressure | Particulates in sample; Aggregated proteins | Clarify sample by centrifugation or filtration prior to loading [20] |
| Poor Reproducibility | Inconsistent sample preparation; Variable flow rates; Column degradation | Standardize sample prep protocol; Control flow rate precisely; Monitor column performance with controls [22] [23] |
## Quantitative Data on Depletion Performance
The following table summarizes performance data for representative commercial depletion products, as derived from manufacturer information. Always consult the specific product datasheet for the most accurate and complete data.
Table 1: Comparison of Representative Abundant Protein Depletion Products
| Product Name | Proteins Depleted | Sample Volume | Processing Time | Depletion Efficiency |
|---|---|---|---|---|
| Pierce Albumin Depletion Kit [20] | Albumin | 10–50 µL | 20–30 min | >95% Albumin |
| High-Select HSA/Immunoglobulin Depletion Spin Columns [20] | Albumin, IgG, IgA, IgM, IgD, IgE | 10 µL or 100 µL | 5–10 min | >95% Albumin & IgGs |
| High-Select Top14 Abundant Protein Depletion Spin Columns [20] | Albumin, IgG, IgA, IgM, IgD, IgE, α1-Acid glycoprotein, Fibrinogen, Haptoglobin, α1-antitrypsin, α2-macroglobulin, Transferrin, Apolipoprotein A-I | 10 µL or 100 µL | 5–10 min | >99% of all 14 proteins |
## Experimental Protocol: Immunoaffinity Depletion Using Spin Columns
This protocol provides a general workflow for depleting abundant proteins from serum using pre-packed spin columns. Always adhere to the manufacturer's specific instructions.
Principle: Antibodies against specific abundant proteins are immobilized on a resin. When serum is passed through the column, these proteins are bound and retained, while the depleted serum (flow-through) is collected for downstream analysis.
Materials & Reagents:
- Pre-packed Depletion Spin Column (e.g., targeting top 2 or top 14 proteins) [20]
- Serum or plasma sample
- Equilibration/Binding Buffer (typically provided or specified)
- Wash Buffer (typically provided or specified)
- Collection Tubes
- Microcentrifuge
Workflow:
Step-by-Step Procedure:
- Equilibration: Place the spin column in a provided collection tube. Apply the recommended volume of equilibration buffer to the column. Centrifuge for the specified time and speed (e.g., 1 minute at 1000 × g). Discard the flow-through.
- Sample Loading: Apply the clarified serum sample (volume not to exceed column capacity) to the center of the resin bed. Centrifuge as in Step 1. The flow-through from this step contains the depleted serum and should be collected.
- Washing: To maximize recovery, apply wash buffer to the column and centrifuge again. Combine this flow-through with the one from Step 2. This is your final depleted serum sample.
- (Optional) Elution: If desired, the bound abundant proteins can be recovered by applying an elution buffer (often a low-pH buffer) and centrifuging. The flow-through from this step contains the eluted abundant proteins.
- Sample Analysis: The depleted serum (flow-through from steps 2 and 3) is now ready for downstream processing, such as digestion for mass spectrometry or direct analysis via biosensors [20].
## The Scientist's Toolkit: Key Research Reagent Solutions
Table 2: Essential Materials for Abundant Protein Depletion
| Item | Function | Example / Note |
|---|---|---|
| Immunoaffinity Spin Columns | Selective removal of target abundant proteins via antibody-antigen binding. | Available in various formats (e.g., albumin-only, top 6, top 12/14 proteins) [20]. |
| Binding/Wash Buffers | To maintain optimal pH and ionic strength for specific binding while minimizing non-specific interactions. | Often supplied with kits; composition is critical for performance [2] [22]. |
| BCA Protein Assay Kit | To estimate total protein concentration in the original and depleted serum, helping to gauge depletion efficiency. | A standard colorimetric method [20]. |
| ELISA Kits | To quantitatively measure the concentration of specific abundant proteins (e.g., Albumin, IgG) before and after depletion. | Used for precise verification of depletion efficiency for individual proteins [20]. |
| Surfactants (SDS, CTAB) | To mitigate non-specific adsorption on equipment and resins by blocking charged functional groups. | Use with caution as they can interfere with downstream MS; must be compatible with the protocol [7]. |
In research involving complex biological samples, such as serum, non-specific adsorption (NSA) of interfering biomolecules to experimental surfaces (e.g., biosensors, microplates, and microscopy slides) is a pervasive challenge. NSA leads to elevated background noise, false-positive signals, reduced sensitivity, and poor reproducibility, which can severely compromise data integrity. Surface passivation—the process of coating surfaces to minimize these unwanted interactions—is therefore a critical step in experimental design. Among the most common passivating agents are Bovine Serum Albumin (BSA) and normal sera (e.g., goat, donkey). However, their effectiveness is not universal and depends heavily on the specific experimental conditions. This technical support center provides troubleshooting guides and FAQs to help researchers optimize the use of these blocking agents to achieve superior results in their studies.
Frequently Asked Questions (FAQs)
Q1: Why does non-specific binding still occur even after I've blocked my surface with BSA? NSA can persist for several reasons related to the BSA itself and the surface:
- BSA Quality and Type: Commercial BSA preparations vary. "Fatted" BSA (containing bound fatty acids) is more conformationally stable but may form a less effective, looser passivation layer compared to "defatted" BSA, which unfolds more readily on surfaces to create a tighter, more protective barrier [24].
- Incomplete Coverage: The BSA layer may not fully cover all surface chemistries, leaving gaps for proteins to adsorb.
- Insufficient Concentration or Time: The blocking step may have been too short or used a sub-optimal BSA concentration.
- Nature of the Interfering Molecule: Certain molecules or fluorophores (e.g., Atto 647N) have a high intrinsic affinity for hydrophobic or surfactant-coated surfaces and may require specialized passivation strategies [25].
Q2: What is the fundamental difference between using BSA and normal serum for blocking? The choice hinges on the primary source of interference in your experiment:
- Bovine Serum Albumin (BSA): This is a single-protein blocker. It works by forming a physical barrier on the surface, reducing available sites for non-specific binding. It is highly effective for general purpose blocking but may not be sufficient for all applications [1].
- Normal Serum: This is a multi-component blocker. It contains a mixture of proteins, including albumin and immunoglobulins (antibodies). It is particularly crucial for immunoassays (like ELISA or immunohistochemistry) because the immunoglobulins in the serum will bind to any remaining, non-specific reactive sites on the surface that might otherwise capture the primary or secondary antibodies used in your assay, thereby preventing false positives.
Q3: My single-molecule fluorescence experiment requires imaging in a high-concentration of labeled proteins. What are my passivation options? Standard PEG-passivated surfaces often fail under high protein concentrations (> low nM). An advanced solution is the DDS-Tween-20 (DT20) surface. This method uses a dimethyldichlorosilane (DDS)-coated surface with adsorbed biotinylated BSA, followed by a self-assembled layer of the surfactant Tween-20. This combination has been shown to reduce non-specific binding of proteins and nucleic acids by up to 30-fold compared to PEG surfaces, while preserving biomolecular activity [25].
Q4: How does the purity and preparation of BSA affect its blocking performance? The purification method of BSA significantly impacts its conformational flexibility and, consequently, its performance as a blocking agent. As detailed in the table below, fatty acid-free (defatted) BSA is generally superior for forming high-quality antifouling coatings [24].
Table: Impact of BSA Type on Passivation Coating Properties
| BSA Type | Fatty Acid Content | Conformational Stability | Adsorption & Coating Properties | Recommended Use |
|---|---|---|---|---|
| Fatted BSA | Contains fatty acids (e.g., from heat-shock fractionation) | Higher | Forms less mass, more viscoelastic, and less tightly packed adlayers. | General blocking where extreme NSA is not a concern. |
| Defatted BSA | Fatty acids removed (e.g., via charcoal treatment) | Lower | Unfolds more on surfaces; forms greater mass, more rigid, and tightly packed coatings. | Superior for high-performance antifouling applications on flat surfaces and nanoparticles [24]. |
Troubleshooting Guide: Common Problems and Solutions
Table: Troubleshooting Non-Specific Adsorption Issues
| Problem | Potential Causes | Recommended Solutions |
|---|---|---|
| High Background in Immunoassays | 1. Secondary antibody binding non-specifically.2. Inadequate blocking of reactive sites. | 1. Use normal serum from the host species of the secondary antibody as a blocker.2. Optimize the concentration and incubation time of the blocking serum [1]. |
| Non-Specific Binding in Single-Molecule Studies | 1. Standard PEG passivation is insufficient for the protein concentration used.2. Sticky biomolecules or fluorophores. | 1. Implement the DT20 passivation method [25].2. Avoid fluorophores known to interact with surfactant layers (e.g., Atto 647N, non-sulfonated Cy3) [25]. |
| Poor Reproducibility & Broad Peak Shapes in Chromatography | Analyte loss due to non-specific adsorption to system surfaces. | 1. Use dedicated, low-NSA columns.2. Incorporate passivation steps (e.g., with BSA) into the conditioning protocol [26]. |
| Variable Passivation Performance with BSA | Inconsistent BSA sources or types between experiments. | Standardize on a single, high-quality source of defatted BSA for critical applications to ensure consistent conformational and adsorption properties [24]. |
Detailed Experimental Protocols
Protocol 1: Advanced Surface Passivation with DDS-Tween-20 (DT20) for Single-Molecule Imaging
This protocol describes how to create a surface that resists non-specific binding far more effectively than traditional PEGylated surfaces [25].
Principle: A surface is first made hydrophobic with dimethyldichlorosilane (DDS). Biotinylated BSA is then non-specifically adsorbed to this surface. Finally, the surfactant Tween-20 self-assembles onto the DDS-coated surface, creating a highly effective passivation layer. The biotinylated BSA allows for specific tethering of biomolecules via biotin-NeutrAvidin interaction.
Diagram: DT20 Surface Passivation Workflow
Materials:
- DDS (dimethyldichlorosilane): Creates the base hydrophobic layer.
- Biotinylated BSA: Provides biotin ligands for specific tethering.
- Tween-20: Forms the primary passivation layer.
- NeutrAvidin: Links the biotin on the surface to biotinylated molecules of interest.
- Anhydrous toluene: Solvent for DDS.
Procedure:
- Surface Cleaning: Thoroughly clean glass coverslips using a vigorous protocol (e.g., piranha etch or plasma cleaning) to ensure a pristine, hydrophilic surface.
- DDS Coating: Incubate the clean, dry coverslips in a 5% (v/v) solution of DDS in anhydrous toluene for 30 minutes. This silanization step creates a hydrophobic surface.
- Rinsing: Rinse the DDS-coated coverslips extensively with toluene, followed by methanol, and then dry under a stream of nitrogen or argon.
- BSA Adsorption: Incubate the DDS-coated surfaces with a solution of biotinylated BSA (e.g., 0.5 mg/mL in PBS) for 10 minutes.
- Tween-20 Passivation: Without rinsing off the BSA solution, add Tween-20 directly to the solution to a final concentration of 0.2% (v/v). Continue the incubation for another 10 minutes. This allows Tween-20 to self-assemble onto the DDS-coated areas.
- Final Wash: Rinse the prepared DT20 surface with a suitable buffer (e.g., PBS) to remove excess reagents. The surface is now ready for incubation with NeutrAvidin and subsequent tethering of biotinylated biomolecules.
Protocol 2: Optimizing BSA-based Nanoparticles for Enhanced Drug Delivery
This protocol outlines the preparation of BSA nanogels (BSA-NGs) via the desolvation method, optimized for applications like nasal drug delivery where mucoadhesion and controlled release are desired [27].
Principle: The gradual addition of a desolvating agent (ethanol) to an aqueous BSA solution causes protein denaturation and coacervation, leading to the formation of nanoparticles. Subsequent stabilization (e.g., with glutaraldehyde) creates a nanogel.
Materials:
- Bovine Serum Albumin (BSA): The matrix-forming polymer.
- Ethanol (Absolute): Desolvating agent.
- Purified Water: Solvent for the initial BSA solution.
- Glutaraldehyde (or similar crosslinker): For gelation and stabilization of nanoparticles.
Procedure:
- Preparation: Dissolve BSA in purified water to create a 20% (w/v) stock solution.
- Desolvation: Under constant stirring (e.g., magnetic stirrer at 500 rpm), slowly add absolute ethanol to the BSA solution. The ratio of BSA solution to ethanol is critical for determining the final nanoparticle size and properties. For example, a formulation of 1.0 mL BSA (20%), 1.2 mL ethanol, and 0.1 mL purified water yielded particles with a Z-average of ~138 nm and a PdI of 0.418 [27].
- Gelation/Cross-linking: Introduce a cross-linking agent, such as glutaraldehyde, to the turbid mixture to solidify the nanoparticles. Allow the reaction to proceed for several hours.
- Purification: Purify the resulting BSA-NGs by centrifugation, washing, and resuspension in the desired buffer.
- Characterization: Characterize the nanoparticles for size (Z-average), polydispersity (PdI), and zeta potential using dynamic light scattering (DLS).
The Scientist's Toolkit: Essential Research Reagents
Table: Key Reagents for Surface Passivation and Their Functions
| Reagent | Function / Key Property | Application Notes |
|---|---|---|
| Defatted BSA | Blocking agent with high conformational flexibility for tight surface packing [24]. | Superior for creating high-performance antifouling coatings on sensors and nanoparticles. |
| Normal Serum | Multi-component blocker containing immunoglobulins to prevent antibody cross-reactivity [1]. | Essential for immunoassays. Must be from a species that matches the secondary antibody host. |
| Tween-20 | Non-ionic surfactant that forms a self-assembled passivation layer [25]. | Core component of the high-performance DT20 surface. |
| Polyethylene Glycol (PEG) | Polymer used for passivation; creates a hydrophilic, neutral barrier [25]. | A common standard, but can be outperformed by newer methods like DT20, especially at high analyte concentrations. |
| Dimethyldichlorosilane (DDS) | Silane used to create a hydrophobic surface foundation [25]. | The first step in the DT20 surface preparation protocol. |
| Casein | Milk-derived protein blocker; effective for many immunoassays [1]. | A common alternative to BSA, often found in commercial blocking buffers. |
Visualizing the Passivation Selection Logic
Choosing the right passivation strategy is critical. The following flowchart provides a logical guide for selecting an appropriate method based on your experimental goals.
Diagram: Passivation Strategy Selection Logic
What is non-specific adsorption (NSA) and why is it a critical issue in biosensing? Non-specific adsorption (NSA), often referred to as biofouling, is the undesirable adhesion of molecules (like proteins, cells, or other biomolecules) to surfaces beyond the intended specific binding events. When working with complex biological samples such as serum or cell lysate, these surfaces are exposed to a high concentration of interfering proteins (e.g., 60–80 mg mL⁻¹ in blood) [28]. NSA leads to elevated background signals, false positives, reduced sensitivity and selectivity, and compromised reproducibility of biosensors and assays [1]. Effectively managing NSA is therefore a foundational requirement for successful research and development in diagnostics and drug development.
Coating Technologies: Mechanisms and Performance
This section details the core antifouling materials, their modes of action, and a direct comparison of their performance in realistic conditions.
Polyethylene Glycol (PEG) and Derivatives
How do PEG-based coatings prevent fouling? PEG creates a hydrated, neutral, and dynamic physical barrier on surfaces. Its anti-fouling properties are primarily attributed to the steric repulsion mechanism and the formation of a hydration layer [29]. The flexible PEG chains, when densely packed, occupy space and physically prevent foulants from reaching the surface. Furthermore, their hydrophilic nature binds water molecules, creating a thermodynamic barrier that is energetically unfavorable for proteins to penetrate or adsorb onto [30] [29]. PEG is often used in grafted copolymer structures, such as PLL-g-PEG, which electrostatically adsorbs to negatively charged surfaces, presenting a dense brush of PEG chains to the solution [30] [29].
Hydrogel Dextran
What is the role of dextran in antifouling applications? Dextran is a hydrophilic polysaccharide that can form a 3D hydrogel matrix on sensor surfaces. This hydrogel structure is highly hydrated, creating a physical and energetic barrier that resists the diffusion and adsorption of proteins [30]. The porous nature of the dextran matrix also allows for high-capacity immobilization of biorecognition elements (e.g., antibodies), making it a popular choice for platforms like Surface Plasmon Resonance (SPR) biosensors. Its effectiveness has been demonstrated in comparative studies against other coatings [6].
Surface-Initiated Polymerization (SIP)
Why is Surface-Initiated Polymerization considered a promising advanced coating? SIP is a technique for growing dense, well-defined polymer brushes directly from a surface. This method allows for precise control over the brush thickness, density, and composition. In comparative studies, SIP-produced surfaces have demonstrated superior performance, showing high sensitivity and the minimum non-specific adsorption of cell lysate and serum among the tested platforms, including PEG and dextran [6]. The dense, covalently attached polymer brush layer presents a formidable steric and hydrated barrier to foulants, making it a strong candidate for a universal biosensor platform.
Table 1: Comparative Performance of Antifouling Coatings in Complex Media
| Coating Type | Mechanism of Action | Performance in Serum/Cell Lysate | Key Advantages | Key Limitations |
|---|---|---|---|---|
| PEG/PLL-g-PEG | Steric repulsion, Hydration layer | Effective reduction of NSA [30] [29] | Well-established, commercially available, highly effective | Can be susceptible to oxidative degradation |
| Dextran (Hydrogel) | 3D Hydration, Size exclusion | Low NSA; good for biosensor platforms [6] | High loading capacity for bioreceptors | Hydrogel thickness can reduce sensitivity in some optical sensors [28] |
| SIP-based Brushes | Dense polymer brush, Steric barrier | Minimum NSA and high sensitivity [6] | Tunable thickness/density, high stability | More complex surface fabrication required |
Experimental Protocols for Coating Evaluation
A robust experimental workflow is essential for developing and validating antifouling surfaces. The diagram below outlines a general protocol for preparing and testing these coatings.
Diagram 1: Workflow for preparing and testing antifouling coatings.
Detailed Protocol: Evaluating Coatings via SPRi and MALDI-TOF MS
This protocol is adapted from a comparative study that used Surface Plasmon Resonance imaging (SPRi) and mass spectrometry to evaluate different coatings [6].
Objective: To quantify and compare the non-specific adsorption of human serum and cell lysate on PEG, dextran, and SIP-modified gold biosensor surfaces.
Materials Needed:
- Sensor Chips: Gold-coated SPRi chips.
- Chemicals:
- Poly-L-lysine-graft-polyethylene glycol (PLL-g-PEG)
- Dextran hydrogel coating reagents
- SIP initiator and monomer solutions
- Human serum (e.g., fetal bovine serum)
- Cell lysate (prepared from relevant cell lines)
- Running buffer (e.g., phosphate-buffered saline, PBS)
- Equipment:
- SPRi instrument
- FTIR Spectrometer
- MALDI-TOF/TOF Mass Spectrometer
- Microfluidic flow cells
Procedure:
- Surface Fabrication & Confirmation:
- Functionalize separate gold SPRi chips with PEG (via PLL-g-PEG adsorption), hydrogel dextran, and SIP brushes according to established synthetic procedures.
- Confirm successful surface modification using Fourier Transform Infrared Spectroscopy (FTIR) to verify the presence of characteristic chemical bonds [6].
SPRi Measurement of NSA:
- Mount the coated chips in the SPRi instrument.
- Prime the system with running buffer at a constant flow rate (e.g., 5-10 µL/min) until a stable baseline is achieved.
- Introduce the complex sample (undiluted serum or cell lysate) over the sensor surface for a fixed period (e.g., 15-30 minutes).
- Monitor the SPRi response in real-time. The change in reflectivity (ΔRU) is directly proportional to mass adsorption on the surface.
- Switch back to running buffer to wash away loosely bound molecules. The remaining signal corresponds to irreversibly adsorbed material (NSA).
- Compare the final NSA response units (ΔRU) across the different coatings. A lower signal indicates better antifouling performance.
Post-Analysis via MALDI-TOF MS:
- After the SPRi run, carefully remove the sensor chips.
- Recover the proteins non-specifically adsorbed to each coating type using a suitable elution method.
- Analyze the eluted proteins using MALDI-TOF/TOF Mass Spectrometry.
- This step identifies the specific proteins in the serum or lysate that adhered to each surface, providing a molecular-level understanding of the fouling process [6].
Expected Outcome: The study following this methodology found that while all "non-fouling" surfaces showed some level of NSA, SIP-based coatings consistently exhibited the lowest ΔRU signal and thus the best performance, followed by dextran and PEG [6].
Troubleshooting Guide & FAQs
Q1: My antifouling coating shows good performance in buffer but fails in 100% serum. What could be the reason? A: This is a common challenge. The complexity and high protein concentration of serum are far more demanding.
- Insufficient Coating Density/Thickness: The polymer brush or hydrogel layer may not be dense or thick enough to effectively shield the underlying "sticky" substrate from the diverse proteins in serum. Consider optimizing your synthesis to increase grafting density for SIP or the cross-linking for dextran [6] [29].
- Coating Degradation: PEG, in particular, is susceptible to oxidative degradation. Check the stability of your coating by analyzing it after exposure to serum. Switching to more stable alternatives like zwitterionic polymers might be necessary [28].
- Sample Variability: Be aware that serum from different donors can have varying protein compositions, which may lead to different NSA profiles. Validate your coating using pooled sera from multiple donors [28].
Q2: How can I functionalize my antifouling coating without compromising its properties? A: Incorporating functional groups during the coating synthesis is key.
- Use Functionalized Copolymers: For PLL-g-PEG, use a commercial variant that includes a fraction of PLL-g-PEG-biotin (e.g., 0.5-1%). This allows you to use a streptavidin bridge to immobilize any biotinylated antibody while the majority of the surface remains non-fouling [30].
- Design SIP with Active Handles: Incorporate a small percentage of functional monomers (e.g., containing carboxyl or amine groups) during the SIP process. These groups can be activated later for biomolecule conjugation without significantly altering the antifouling properties of the brush [6].
Q3: Why is the signal from my specific target binding event still low, even with a good antifouling coating? A: This could be due to several factors:
- Steric Hindrance: The antifouling layer might be so dense that it physically blocks the access of your large target analyte (or the immobilized bioreceptor) to its binding partner. Optimize the thickness of the coating to find a balance between fouling resistance and binding efficiency [28].
- Inadequate Bioreceptor Immobilization: The method used to attach your antibody or aptamer might be inefficient, leading to low capture capacity. Ensure your functionalization protocol is optimized for high yield and proper orientation of the bioreceptor.
Table 2: Research Reagent Solutions for Antifouling Experiments
| Reagent/Material | Function in Experiment | Example Use Case |
|---|---|---|
| PLL-g-PEG | Pegylated polyelectrolyte for easy coating | One-step adsorption onto negatively charged surfaces (e.g., plasma-treated PDMS or metal oxides) to create a PEG brush [30] [29]. |
| PLL-g-PEG-Biotin | Functionalized pegylated polyelectrolyte | Co-adsorbed with PLL-g-PEG to introduce biotin groups for subsequent streptavidin and biotinylated antibody immobilization [30]. |
| Dextran-based matrix | Hydrogel coating for biosensors | Forming a 3D, hydrophilic matrix on SPR sensor chips to resist fouling and provide a scaffold for ligand immobilization [6]. |
| SI-ATRP Initiator | Molecule to start Surface-Initiated Atom Transfer Radical Polymerization (SI-ATRP) | Grafted onto a gold surface to initiate the growth of polymer brushes (e.g., PEG-like or zwitterionic) via SIP [6]. |
| Human Serum/Fetal Bovine Serum | Complex biological challenge medium | Used undiluted or diluted to test the antifouling efficacy of coatings under realistic conditions [6] [28]. |
The battle against non-specific adsorption in complex samples is ongoing. While PEG remains a widely used and effective standard, and dextran hydrogels offer excellent bioreceptor loading capacity, advanced coatings like those created through Surface-Initiated Polymerization are showing superior performance in head-to-head studies [6]. The future of this field lies in the development of even more robust and smart materials, such as zwitterionic polymers and hybrid coatings. The integration of high-throughput screening and machine learning will further accelerate the discovery and optimization of next-generation antifouling surfaces, ultimately enabling more reliable and sensitive diagnostic and research tools [2].
FAQs: Tackling Non-Specific Adsorption in Serum Samples
Q1: What is the primary cause of non-specific adsorption (NSA) when using MIPs in complex serum samples? Non-specific adsorption occurs due to unwanted physical and chemical interactions between the biosensing interface and various components in the serum matrix. These are primarily driven by hydrophobic interactions, electrostatic forces, hydrogen bonding, and van der Waals forces [2]. In serum, which is rich in proteins, fats, and other biomolecules, these interactions can lead to the fouling of the MIP surface, masking the specific binding sites for your target analyte and compromising the sensor's signal and selectivity [2].
Q2: How can surfactant modification help reduce NSA in my MIP-based assay? Surfactants can significantly suppress NSA by interfering with the weak, non-covalent forces that cause it [31]. Their amphipathic nature allows them to interact with both the polymer surface and the hydrophobic components of the sample matrix. Using surfactants in sub-micellar concentrations (below the critical micellar concentration) in your binding buffer is crucial. Ionic surfactants (e.g., SDS, CTAB) generally have a stronger depressive effect on NSA than non-ionic surfactants (e.g., Tween 20), but they may also reduce the specific binding affinity of the MIP for its template [31].
Q3: What are "dummy templates" and when should I use them? A dummy template is a structural analog of your target molecule that is used during the MIP synthesis instead of the actual target. This strategy is particularly valuable when the target molecule is toxic, expensive, or unstable during the polymerization process [32]. It also entirely avoids the problem of "template leakage," where residual template molecules leach out of the MIP during application, causing false positives and inaccurate quantification [32].
Q4: My MIPs lack consistency between batches. How can I improve reproducibility? Reproducibility is a common challenge in MIP synthesis. To improve it, focus on standardizing these key parameters [32]:
- Template-Monomer Complexation: Ensure consistent pre-polymerization conditions (solvent, time, temperature) for complex formation.
- Polymerization Method: Shift from traditional bulk polymerization to methods that yield more uniform particles, such as precipitation polymerization or surface imprinting [32].
- Cross-linker Density: Maintain a high and consistent cross-linker-to-monomer ratio to create a rigid polymer network that "locks" the recognition sites in place [32].
Q5: Are there sustainable alternatives for creating MIPs? Yes, the field is moving towards greener materials. Biomass-based MIPs (bio-based MIPs) are gaining attention. These utilize sustainable resources like polysaccharides (e.g., chitosan, cellulose) or biomass-derived carbon as base materials [33]. These polymers are not only environmentally friendly but can also offer abundant active functional groups for imprinting [33].
Troubleshooting Guides
Table 1: Troubleshooting Low Selectivity in Complex Serum
| Observed Problem | Potential Cause | Recommended Solution |
|---|---|---|
| High background signal in serum | Hydrophobic interactions with serum proteins | Add a non-ionic surfactant like Tween 20 (0.01-0.1% v/v) to the binding and washing buffers [31] [2]. |
| Poor differentiation from structural analogs | Inadequate complementarity of binding sites | Re-optimize the monomer-to-template ratio; use a more specific functional monomer (e.g., 4-vinylpyridine for acidic targets) [32]. |
| Signal degradation over multiple uses | Fouling from accumulated serum components | Implement a stringent regeneration protocol using a wash with a low-percentage ionic surfactant (e.g., 1-5 mM SDS) or an acidic/basic solution [31]. |
| Inconsistent imprinting factor | Use of a polar solvent that disrupts key interactions | Switch to a porogenic solvent with a lower dielectric constant (e.g., toluene or chloroform) to strengthen hydrogen bonding during polymerization [32]. |
Table 2: Troubleshooting Surfactant Modification Protocols
| Observed Problem | Potential Cause | Recommended Solution |
|---|---|---|
| Severe loss of specific binding signal | Surfactant concentration is too high, disrupting template binding | Titrate the surfactant concentration. Ensure it is below the Critical Micellar Concentration (CMC) and use the lowest effective dose [31]. |
| Non-ionic surfactant is ineffective | Hydrophobic NSA is not the primary issue; electrostatic interactions may dominate | Test a charged surfactant or a mixed surfactant system. Alternatively, adjust the pH or ionic strength of the buffer to shield electrostatic forces [2]. |
| Signal instability or drift | Surfactant interacting with the transducer or detection chemistry | Characterize the surfactant's effect on the full sensor system. Consider using a different surfactant type (e.g., switch from ionic to non-ionic) or a different antifouling coating [2]. |
Experimental Protocols
Protocol 1: Optimizing Surfactant-Enhanced Binding Buffer for Serum Samples
This protocol outlines a method to incorporate surfactants into your binding assay to minimize NSA.
Materials:
- MIP particles or sensor
- Target analyte
- Surfactants: Tween 20 (non-ionic), SDS (anionic), CTAB (cationic)
- Binding buffer (e.g., phosphate or acetate buffer)
- Complex sample (e.g., diluted serum)
Method:
- Prepare Surfactant Stocks: Create concentrated stock solutions of each surfactant in your binding buffer.
- Spike Binding Buffers: Add varying, sub-micellar amounts of each surfactant to separate aliquots of binding buffer. For example, prepare a series with Tween 20 (0.01%, 0.05%, 0.1% v/v) and SDS/CTAB (0.1 mM, 0.5 mM, 1.0 mM).
- Binding Assay: Incubate a fixed amount of your MIP with a known concentration of the target analyte spiked into the surfactant-containing buffers and a serum sample.
- Quantify Binding: After incubation and separation, measure the amount of bound target (e.g., via HPLC, spectrophotometry).
- Calculate Performance Metrics: For each condition, calculate the binding affinity and the imprinting factor (IF = Keq(MIP)/Keq(NIP)) [31].
- Select Optimal Condition: Choose the surfactant type and concentration that yields the highest imprinting factor and recovery from serum, indicating strong specific binding with minimal NSA.
Protocol 2: Solid-Phase Extraction (SPE) of Serum Contaminants Using MIPs
This protocol uses a MIP as a selective sorbent to clean up serum samples before analysis.
Materials:
- MIP-packed SPE columns
- Control: Non-imprinted polymer (NIP)-packed SPE columns
- Serum samples
- Loading buffer (e.g., water or a weak buffer)
- Wash buffer (e.g., loading buffer with 5-10% acetonitrile)
- Elution solvent (e.g., methanol with 1% acetic acid)
Method:
- Conditioning: Condition the MIP and NIP SPE columns with elution solvent followed by loading buffer.
- Sample Loading: Dilute the serum sample with loading buffer and load it onto the columns.
- Washing: Wash the columns with wash buffer to remove non-specifically bound contaminants. The wash buffer can be optimized with surfactants like Tween 20 to enhance the removal of NSA components [31] [32].
- Elution: Elute the specifically bound target analyte with a strong elution solvent.
- Analysis: Analyze the eluate using your preferred detection method (e.g., LC-MS). Compare the chromatograms from the MIP and NIP columns. A clean chromatogram with a strong target peak from the MIP, compared to a noisy one from the NIP, demonstrates effective selective extraction and NSA reduction.
Workflow and Signaling Pathways
The Scientist's Toolkit
Table 3: Key Research Reagent Solutions
| Reagent | Function/Explanation | Application Note |
|---|---|---|
| Tween 20 | A non-ionic surfactant used to block hydrophobic binding sites on the MIP and plasticware, reducing NSA of proteins and lipids from serum [31] [2]. | Typically used at 0.01-0.1% v/v in buffers. Has a milder effect on specific binding compared to ionic surfactants [31]. |
| SDS (Sodium Dodecyl Sulfate) | An anionic surfactant with a strong depressive effect on NSA. Useful as a regeneration agent to clean MIPs between uses [31]. | Use at low, sub-micellar concentrations (e.g., 1-5 mM) to avoid destroying the specific binding cavities of the MIP [31]. |
| 4-Vinylpyridine (4VP) | A common basic functional monomer that interacts with acidic or electron-deficient template molecules via hydrogen bonding or ionic interactions [32]. | Ideal for imprinting acidic targets like phenoxyacid herbicides or certain pharmaceuticals [32]. |
| EGDMA (Ethylene Glycol Dimethacrylate) | A cross-linker that creates a rigid polymer network, stabilizing the three-dimensional shape of the imprinted cavities [32]. | A high cross-linker ratio (70-90%) is typically used to ensure cavity stability and MIP reusability [32]. |
| Dummy Template | A safer or more stable structural analog of the target molecule, used to create specific cavities while avoiding template leakage issues [32]. | Crucial for the imprinting of toxic molecules or when ultimate detection sensitivity is required. |
Troubleshooting High Background and Optimizing Your Protocol
Diagnosing the Source of Unacceptable Background Signals
Troubleshooting Guide: Identifying Causes of High Background
This guide helps you systematically identify and resolve the common causes of unacceptable background signals in immunoassays using complex serum samples.
Problem: High background noise, abnormal signals in negative controls, or low signal-to-noise ratio
| Symptom | Possible Cause | Diagnostic Steps | Solution |
|---|---|---|---|
| High signals across all wells, including negatives | Incomplete blocking [34] [35] | Check blocking buffer concentration and incubation time. | Use a robust blocking agent (e.g., 5% BSA); extend blocking to at least 1 hour [34] [35]. |
| Inadequate washing [35] | Review wash cycle number and duration. | Increase to 3-5 wash cycles; ensure wells are filled completely and buffer sits for 30s before aspiration [35]. | |
| High background in serum samples | Interference from heterophilic antibodies or Rheumatoid Factor (RF) [35] | Test assay with and without spiked normal serum. | Pre-treat samples with 10% normal serum from the same species as the secondary antibody to block interference [35]. |
| Non-specific electrostatic interactions [36] | Analyze antibody physicochemical properties; check for interactions with charged polymers like DNA [36]. | Select antibodies with lower positive charge patches; include non-ionic detergents in buffers [36]. | |
| Non-specific binding to plate | Antibody cross-reactivity [35] | Run assay with monoclonal vs. polyclonal antibodies. | Switch to high-specificity monoclonal antibodies or use cross-adsorbed secondary antibodies [35]. |
| Hydrophobic interactions with plate [35] | Compare signals on standard polystyrene vs. hydrophilic-treated plates. | Switch to hydrophilic-treated plates (e.g., PVDF) for proteins with strong hydrophobic interactions [35]. | |
| High background in luminescence/fluorescence assays | Compound fluorescence or assay interference [37] | Include a pre-read step before adding detection reagents. | Use time-resolved fluorescence (TRF) or red-shifted fluorophores; add non-ionic detergent to reduce aggregation [37]. |
Frequently Asked Questions (FAQs)
Q1: Our assay background was acceptable with buffer but became unacceptable when testing mouse serum samples. What is the most likely cause? A1: Complex biological samples like serum introduce specific interferents. The most common causes are:
- Heterophilic Antibodies: These are endogenous antibodies in the test sample that can bridge the capture and detection antibodies, even in the absence of the target antigen, leading to false-positive signals [35].
- Rheumatoid Factor (RF): An autoantibody that can bind to the Fc region of assay antibodies, also creating a bridge and causing high background [35].
- Charge Interactions: Therapeutic or endogenous antibodies with positive surface charges can form nonspecific interactions with negatively charged biomolecules in serum, such as DNA or insulin [36].
- Solution: Pre-treat samples with 10% normal serum from the same species as the secondary antibody to block these interferents [35].
Q2: We've optimized our protocol, but background remains high. Could the antibodies themselves be the problem? A2: Yes. Antibody developability is a critical factor. Some antibodies, even clinical-stage ones, have inherently poor biophysical properties.
- Cause: During affinity maturation, optimization for target binding can sometimes come at the cost of increased nonspecific interactions. This is often driven by charged or hydrophobic patches on the antibody's surface [36].
- Diagnosis: Utilize solution-based microfluidic technologies to characterize the antibody's hydrodynamic radius and effective charge, generating a "nonspecificity fingerprint" [36].
- Solution: If possible, select an alternative antibody clone with a better developability profile, indicated by a lower nonspecificity score in screening assays [36].
Q3: What are the most effective strategies to reduce non-specific adsorption during the assay procedure itself? A3: A multi-pronged approach targeting key steps is most effective.
- Blocking: Do not reduce blocking time. Use a high concentration (e.g., 5% BSA or non-fat dry milk) and ensure adequate incubation (at least 1 hour) to cover all unbound sites on the plate [34] [35].
- Washing: Be meticulous. Insufficient washing is a major culprit. Ensure multiple cycles (3-5) with a buffer containing a non-ionic detergent like Tween-20 to remove unbound reagents effectively [36] [35].
- Reagent Quality: Use cross-adsorbed secondary antibodies to minimize cross-reactivity, and ensure enzyme conjugates are fresh and active to prevent spurious signal generation [35].
The Scientist's Toolkit: Research Reagent Solutions
| Item | Function & Application | Key Consideration |
|---|---|---|
| BSA (Bovine Serum Albumin) | Standard blocking agent to cover unbound sites on the microplate surface, reducing non-specific protein adsorption [34] [35]. | Use at least 1-2 hours at room temperature; 5% solution is common for robust blocking [35]. |
| Non-ionic Detergent (e.g., Tween-20) | Added to wash buffers to disrupt hydrophobic and electrostatic interactions, effectively washing away unbound reagents [36] [35]. | Typical concentration is 0.01-0.1%; critical for reducing aggregation-based inhibition and background [36] [37]. |
| Cross-Adsorbed Secondary Antibodies | Secondary antibodies that have been purified to remove antibodies that could cross-react with proteins from other species. | Essential for minimizing cross-reactivity in complex samples; use Fab fragment antibodies for even higher specificity [35]. |
| Normal Serum | Used as a blocking agent in the sample buffer to compete with heterophilic antibodies and other serum interferents. | Should be from the same species as the secondary antibody (e.g., 10% normal goat serum if using a goat-anti-mouse secondary) [35]. |
| Hydrophilic-Treated Plates (e.g., PVDF) | An alternative to polystyrene plates for proteins with strong hydrophobic regions, reducing non-specific adsorption via surface chemistry [35]. | Consider if high background persists despite optimization on standard plates. |
Experimental Protocol: Validating Solutions for Serum Samples
This protocol outlines a step-by-step experiment to diagnose and address the source of high background in complex serum samples.
Objective: To determine if high background is caused by serum interferents and to validate the efficacy of normal serum pre-treatment.
Materials:
- Test serum samples
- Assay buffer (e.g., PBS)
- Normal serum (species-matched to secondary antibody)
- Coated and blocked microplate
- Standard assay reagents (detection antibodies, substrate, etc.)
Method:
- Sample Preparation: Create three sets of samples.
- Group A (Buffer Control): Spike a known negative sample into clean assay buffer.
- Group B (Test Serum): The same negative sample in the test serum matrix.
- Group C (Treated Serum): The negative sample in test serum that has been pre-incubated with 10% normal serum for 30 minutes at room temperature [35].
- Assay Execution: Run your standard assay protocol according to the manufacturer's or your lab's instructions, ensuring all three sample groups are processed simultaneously on the same plate.
- Data Analysis: Compare the background signals (e.g., from negative controls or zero analyte wells) across the three groups.
Interpretation of Results:
- If background in Group B > Group A, serum interferents are likely present.
- If background in Group C is significantly reduced compared to Group B, the interference is confirmed and the normal serum treatment is effective.
- If background remains high in Group C, investigate other causes like insufficient plate blocking or antibody cross-reactivity [35].
Diagnostic Workflow for Background Signals
The diagram below outlines a logical pathway to diagnose the source of high background signals.
In research involving complex serum samples, non-specific adsorption (NSA) presents a significant challenge that can compromise assay accuracy, sensitivity, and reproducibility. NSA occurs when proteins, lipids, or other biomolecules from samples adsorb to assay surfaces through hydrophobic interactions, ionic bonds, or van der Waals forces, leading to high background signals and false positives [1] [2]. Effective blocking—the process of saturating unoccupied binding sites on solid surfaces with inert agents—is therefore imperative for successful experiments such as ELISAs and Western blots, particularly when working with biologically complex matrices like blood, serum, and milk [38] [39] [2].
This guide provides targeted troubleshooting advice to help researchers optimize blocking conditions to minimize NSA, thereby improving the reliability of data generated in drug development and diagnostic applications.
Frequently Asked Questions (FAQs)
What is non-specific adsorption and how does it affect my assay?
Non-specific adsorption (NSA), also called non-specific binding (NSB) or biofouling, refers to the undesirable adhesion of molecules (like proteins from serum samples) to your assay surface or detection components [1] [39]. This occurs via physisorption through hydrophobic forces, electrostatic interactions, and hydrogen bonding [1] [4]. In practice, NSA increases background noise, reduces the signal-to-noise ratio, causes false-positive readings, and can even mask weak specific signals, leading to inaccurate data interpretation [1] [39] [2].
Why is blocking especially critical for assays using serum samples?
Serum is a highly complex matrix containing a high concentration of diverse proteins (such as albumin and immunoglobulins), lipids, and other biomolecules [2]. These components compete with your target analyte for binding sites on the assay surface (e.g., microplate wells or membrane surfaces). Without effective blocking, these serum constituents will adsorb non-specifically, significantly increasing background interference and reducing the assay's ability to accurately detect the specific target [39] [2].
How do I choose between different blocking buffers like BSA, non-fat milk, and casein?
The choice of blocker is system-dependent and involves trade-offs between blocking efficiency, background, and compatibility with your detection system. No single blocking agent is ideal for every situation [38].
- Non-fat milk (e.g., 2-5%): Cost-effective and a common choice for many applications. However, it contains phosphoproteins and biotin, which can interfere with the detection of phosphorylated proteins or streptavidin-biotin systems [38].
- Bovine Serum Albumin (BSA, e.g., 2-5%): A purer protein often preferred for phosphorylated protein detection and biotin-streptavidin systems. It can provide higher sensitivity but may sometimes be a weaker blocker than milk, potentially resulting in higher background for some antibodies [38] [39].
- Casein: An excellent blocking protein known for providing low background. It is a good choice when milk causes high background or interferes with antigen-antibody binding [38].
- Specialized Commercial Blockers: Many proprietary blocking buffers (e.g., protein-based or protein-free formulations) are optimized for specific applications like fluorescent Western blotting, offering low autofluorescence and consistent performance [38] [40].
What is the role of detergents like Tween 20 in blocking and washing?
Detergents like Tween 20 are used in blocking buffers and wash solutions (typically at 0.05%-0.2%) to reduce hydrophobic interactions that drive NSA [38]. They help dissociate weakly bound molecules from surfaces. However, caution is needed as high concentrations (>0.2%) can potentially elute weakly-binding specific antibodies, reducing your target signal [38]. For fluorescent assays, be aware that dried Tween 20 can autofluoresce, so ensure blots are imaged wet or use detergent-free blockers for this step [41] [38].
Troubleshooting Guide
Problem: High Background Signal Across the Entire Membrane or Plate
| Potential Cause | Recommended Solution |
|---|---|
| Insufficient blocking agent concentration or time | Increase blocker concentration (e.g., from 2% to 5%) and/or extend incubation time to a minimum of 1 hour at room temperature. Ensure sufficient buffer volume (≥0.4 mL/cm²) [40]. |
| Ineffective blocking agent for your system | Empirically test alternative blockers. Switch from milk to BSA or a specialized commercial blocker, especially if detecting phosphoproteins or using streptavidin-biotin systems [38] [39]. |
| Inadequate washing | Incorporate Tween 20 (0.05%-0.1%) into wash buffers and increase the number or duration of wash steps post-antibody incubation [38]. |
Problem: High Background in Fluorescent Western Blot
| Potential Cause | Recommended Solution |
|---|---|
| Autofluorescence of blocking agents or detergents | Use blockers specifically formulated for fluorescence (e.g., protein-free or clarified buffers). Filter all buffers to remove particulates. Avoid letting detergents like Tween 20 dry on the membrane [41] [38]. |
| Suboptimal buffer system | For detecting phosphoproteins, use Tris-buffered saline (TBS) instead of phosphate-buffered saline (PBS), as phosphates in PBS can interfere with antibody binding [40]. |
Problem: Weak or No Specific Signal
| Potential Cause | Recommended Solution |
|---|---|
| Over-blocking | The blocker may be masking the antigen or inhibiting the antibody. Reduce the concentration of the blocking agent or switch to a different type (e.g., from milk to BSA) [38]. |
| Antibody incompatibility with blocker | Ensure the primary and secondary antibodies are diluted in the appropriate blocking buffer. For problematic antibodies, pre-test several blockers to find one that preserves specific binding [41] [38]. |
Experimental Optimization Protocols
Protocol for Empirically Testing Blocking Buffers
This systematic approach helps identify the optimal blocking condition for a specific assay [40].
Step-by-Step Method:
- Prepare Samples: Load a serial dilution of your target protein or cell lysate, plus controls, onto a gel. Transfer to a membrane as usual.
- Cut Membrane: After transfer, cut the membrane into several strips, each containing the full sample set.
- Block: Incubate each strip with a different blocking buffer (e.g., 5% BSA, 5% non-fat milk, 2% casein, a commercial specialty blocker) for 1 hour at room temperature with gentle shaking.
- Probe and Detect: Follow standard antibody incubation and detection steps for all strips, using consistent times and reagent dilutions prepared in their respective blocking buffers.
- Analyze: Compare the strips for specific signal strength and background noise to select the best blocker [38] [40].
Protocol for Optimizing Ionic Strength
Ionic strength influences electrostatic interactions that contribute to NSA. This protocol uses a quartz crystal microbalance (QCM) technique to study these interactions [42].
Step-by-Step Method:
- Prepare Solutions: Prepare your buffer (e.g., PBS, pH 7.1-7.4) at different ionic strengths, modified by adding NaCl (e.g., from 0.06 M to 0.56 M) [42].
- Measure Adsorption: Use a QCM or similar biosensor to measure the frequency shift (Δf, related to mass adsorption) and dissipation shift (ΔD, related to viscoelasticity) when the sample in each buffer is introduced to the surface.
- Analyze: The conditions that result in the lowest frequency shift (lowest mass adsorption) indicate the ionic strength that minimizes NSA [42]. The following diagram illustrates this experimental workflow.
The Scientist's Toolkit: Key Research Reagents
| Reagent / Material | Function in Reducing NSA | Key Considerations |
|---|---|---|
| BSA | Inert protein that saturates binding sites on surfaces. | Use high-grade purity; ideal for phosphoprotein detection and streptavidin systems [38] [39]. |
| Non-Fat Dry Milk | Mixed protein solution that effectively blocks unsaturated sites. | Avoid with phosphoprotein detection or biotin systems due to intrinsic contaminants [38]. |
| Casein | Highly effective single-protein blocker derived from milk. | Provides very low background; available as purified sodium salt [38]. |
| Tween 20 | Non-ionic surfactant that reduces hydrophobic interactions. | Use at 0.05-0.2% in buffers; higher concentrations may strip specific antibodies [38]. |
| Low-Binding Consumables | Tubes/plates with surface treatments that minimize analyte adhesion. | Critical for handling molecules prone to NSA (e.g., peptides, nucleic acids) [4]. |
| Specialty Commercial Blockers | Optimized formulations for specific applications (e.g., fluorescence). | Often provide superior performance and consistency but at a higher cost [38] [40]. |
Key Workflow for Systematic Blocking Optimization
The following diagram summarizes the critical decision points and actions in the blocking optimization workflow, integrating the concepts discussed in this guide.
In research involving complex serum samples, non-specific adsorption presents a significant challenge that can compromise data integrity. Unwanted binding of proteins, lipids, or other serum components to solid surfaces leads to increased background noise, reduced assay sensitivity, and inaccurate results. This technical guide provides scientists with strategies for selecting and modifying solid surfaces to minimize non-specific interactions while maintaining target binding capacity, enabling more reliable and reproducible experimental outcomes in serum-based studies.
Surface Selection Guide: Polystyrene & High-Binding Plates
Fundamental Properties and Trade-Offs
The core challenge in surface selection lies in the inherent properties of the materials. Standard polystyrene (PS) surfaces are hydrophobic, promoting protein adsorption primarily through hydrophobic interactions [43]. This makes them prone to non-specific binding from complex samples like serum. In contrast, high-binding plates are specifically treated to enhance protein adsorption capacity, often through surface modification to introduce charged or reactive groups, which can further exacerbate non-specific binding issues.
Decision Framework: A Side-by-Side Comparison
The following table summarizes the key characteristics of each surface type to guide your selection:
| Feature | Standard Polystyrene | High-Binding Plates |
|---|---|---|
| Primary Binding Mechanism | Hydrophobic interactions [43] | Hydrophobic, ionic, and/or covalent interactions |
| Non-Specific Adsorption in Serum | High (due to hydrophobic surface) | Very High (by design) |
| Ideal Application | Samples in simple buffers; low protein concentrations | Targets in purified solutions; high analyte concentration |
| Risk in Serum Samples | High non-specific binding of albumin and other serum proteins [43] | Severe fouling and very high background |
| Mitigation Strategy | Essential surface coating required | Generally not recommended for complex samples |
Diagram: Troubleshooting workflow for high non-specific adsorption in serum samples.
Troubleshooting FAQs: Minimizing Non-Specific Adsorption
Surface Coating and Modification Strategies
Q1: What surface modifications can reduce non-specific adsorption from serum samples on polystyrene plates?
Several coating strategies can transform a polystyrene surface to resist fouling:
- Zwitterionic Polymer Coatings: Surfaces modified with poly(carboxybetaine) (PCB) demonstrate excellent resistance to non-specific protein adsorption and high permeability, allowing for efficient mass transfer while protecting the surface [44]. The electrostatically induced hydration layer formed by zwitterions effectively prevents protein adhesion [45].
- PEGylation: Covalent attachment of polyethylene glycol (PEG) polymers creates a hydrophilic barrier that reduces protein adsorption. A one-step photochemical method using dissolved benzophenone and functionalized PEG can effectively coat polystyrene [46].
- Negatively Charged Films: Creating a dense, self-assembled layer of negatively charged molecules like poly(styrene sulfonic acid) sodium salt (PSS) on the surface can significantly inhibit non-specific adsorption of negatively charged proteins and probes [47].
Q2: How does non-specific adsorption of serum proteins affect my assay, and how can I detect it?
Non-specific adsorption primarily causes two problems:
- Increased Background Signal: Leads to higher noise and poorer signal-to-noise ratio.
- Loss of Analytical Sensitivity: Target molecules may be outcompeted for binding sites by abundant serum proteins like Human Serum Albumin (HSA), which constitutes a large portion of serum protein content [43].
To detect non-specific adsorption, run a negative control (e.g., serum sample without the target analyte) and measure the background signal. A high signal in the negative control indicates significant non-specific binding.
Protocol Optimization and Experimental Design
Q3: Beyond surface coating, what experimental steps can minimize non-specific binding in serum assays?
- Optimized Blocking: Use effective blocking agents like purified BSA, casein, or commercial proprietary blockers. Avoid carriers that contain serum if your sample is serum-based.
- Stringency Washes: Incorporate washes with mild detergents (e.g., Tween-20) to disrupt hydrophobic interactions without denaturing specific bonds.
- Sample Dilution and Composition: Dilute samples in a buffer that minimizes hydrophobic interactions. Adding low concentrations of non-ionic detergents to the dilution buffer can help.
Essential Reagents and Materials
The table below lists key materials referenced in this guide for developing low-fouling surfaces:
| Reagent/Material | Function/Description | Key Reference |
|---|---|---|
| Carboxybetaine Methacrylate (CBMA) | Monomer for creating zwitterionic, anti-biofouling hydrogel coatings. | [44] |
| Poly(styrene sulfonic acid) sodium salt (PSS) | Creates a dense, negatively charged film via self-assembly to reduce non-specific adsorption. | [47] |
| Benzophenone-PEG | Used in a one-step UV photochemical procedure to covalently attach PEG coatings to polymer surfaces. | [46] |
| Poly(2-hydroxyethyl methacrylate) (PHEMA) | A hydrophilic coating material; note it may significantly reduce adsorption rates. | [44] |
| Polyvinylpyrrolidone (PVP) | A hydrophilic polymer used as a coating (e.g., in CytoSorb); can adsorb over 50% of plasma proteins. | [44] |
Advanced Experimental Protocol: Applying a Zwitterionic Coating to Polystyrene Resin
This protocol is adapted from research on creating hemocompatible adsorbents and illustrates a robust method for surface modification [44].
Background and Principle
Encapsulating polystyrene resin in a zwitterionic poly(carboxybetaine) (PCB) hydrogel creates a physical barrier with exceptional anti-biofouling properties. The PCB hydrogel provides high permeability for solute diffusion while effectively resisting the adhesion of proteins and cellular components, making it ideal for complex biological fluids like serum [44].
Step-by-Step Methodology
Materials Required:
- Polystyrene resin microparticles (e.g., H103 resin)
- Carboxybetaine methacrylate (CBMA) monomer
- Crosslinker (e.g., carboxybetaine dimethacrylate)
- Thermal initiator (e.g., Ammonium persulfate, APS)
- Accelerator (e.g., N,N,N',N'-Tetramethylethylenediamine, TEMED)
- Deionized water
- Nitrogen gas source
Procedure:
- Resin Preparation: Wash the pristine polystyrene resin beads thoroughly with ethanol and deionized water. Dry under vacuum.
- Solution Preparation: Prepare an aqueous solution containing the CBMA monomer (e.g., 2.0 M) and the crosslinker (e.g., 2 mol% relative to monomer). Degas the solution with nitrogen for 15-20 minutes to remove dissolved oxygen.
- Initiation: Add the thermal initiator APS (e.g., 0.5 wt%) and the accelerator TEMED (e.g., 0.5 wt%) to the degassed solution and mix gently.
- Encapsulation: Immediately immerse the prepared polystyrene resin beads into the reaction solution. Ensure the resin is fully submerged and dispersed.
- Polymerization: Allow the polymerization to proceed at room temperature for 1-2 hours. The formation of a solid hydrogel around the resin particles indicates a successful reaction.
- Post-treatment: Wash the resulting PCB-encapsulated resin (PCB-H103) extensively with deionized water and PBS to remove any unreacted monomers. Store the final product in PBS at 4°C.
Expected Outcomes and Validation
- Hemocompatibility: The modified resin should show a dramatically reduced hemolysis ratio (e.g., ~0.64% compared to higher percentages for uncoated resin) [44].
- Adsorption Efficiency: The coating should not hinder the adsorption of target toxins. The PCB hydrogel's high permeability can even lead to an adsorption capacity for protein-bound toxins like bilirubin that is 8.3 times higher than that of pristine resin in 100% serum, as it prevents the occlusion of active sites by non-specifically adsorbed proteins [44].
Troubleshooting Guide: FAQs on Reducing Non-Specific Adsorption
This guide addresses common experimental challenges in reducing non-specific adsorption and background interference when working with complex serum samples.
FAQ 1: How can I prevent secondary antibody cross-reactivity in double staining with antibodies from similar species?
A common problem in multiplexed experiments is cross-species reactivity, where a secondary antibody unintentionally binds to a primary antibody from a different species.
- Problem Identification: When using mouse and rat primary antibodies together, a goat anti-mouse secondary may cross-react with the rat primary, and vice versa. This is because mouse and rat immunoglobulins can share antigenic determinants [48].
- Recommended Solution: Sequential Staining with Intermediate Blocking
- Perform stainings sequentially rather than simultaneously. Complete the entire staining cycle (primary antibody, secondary antibody, visualization) for the first antigen before beginning the second [49] [48].
- Incorporate an intermediate blocking step using normal serum from the species of the secondary antibody used in the next cycle. For example, after the first staining cycle, block with normal goat serum before applying the next primary antibody if a goat secondary is used next [48] [50].
- Use pre-adsorbed secondary antibodies specifically cross-adsorbed against immunoglobulins from the potentially cross-reactive species to minimize off-target binding [48].
FAQ 2: What are the most effective surface passivation strategies for biosensors in complex fluids like serum?
Non-specific adsorption (NSA) of proteins and other biomolecules onto biosensor surfaces can mask detection signals and reduce sensitivity.
- Problem Identification: NSA, or biofouling, occurs due to electrostatic, hydrophobic, and van der Waals interactions between the sensor surface and components in complex samples like serum or milk [2].
- Recommended Solutions:
- Zwitterionic Peptide Coatings: Covalently immobilize zwitterionic peptides (e.g., sequences like EKEKEKEKEKGGC) onto the sensor surface. These peptides form a strong, neutral hydration layer that acts as a superior physical and energetic barrier to non-specific adsorption, outperforming traditional polyethylene glycol (PEG) coatings [51].
- Surfactant Modification: For molecularly imprinted polymers (MIPs), electrostatic modification with surfactants like Sodium Dodecyl Sulfate (SDS) or Cetyl Trimethyl Ammonium Bromide (CTAB) can effectively eliminate non-specific binding by reacting with external functional groups on the polymer [7].
- Hybrid Material Coatings: Utilize cross-linked protein films or other hybrid materials designed to create a dense, antifouling layer that resists protein adsorption while maintaining sensor functionality [2].
FAQ 3: How can I optimize immunofluorescence staining to reduce high background signal?
High background noise can obscure specific signals, making data interpretation difficult.
- Problem Identification: High background can stem from various sources, including inadequate blocking, endogenous enzyme activity, over-concentration of antibodies, or non-specific antibody binding [50] [52] [53].
- Recommended Solutions:
- Optimized Blocking: Use a combination of 1-5% Bovine Serum Albumin (BSA) or 10% normal serum from the same species as the secondary antibody for 30-60 minutes. This saturates non-specific binding sites [54] [50] [52].
- Antibody Titration: Always titrate primary and secondary antibodies to find the optimal concentration that provides a strong specific signal with minimal background [52].
- Thorough Washing: Increase the number and duration of washes (e.g., 3 washes for 5 minutes each with an appropriate buffer like TBST) after primary and secondary antibody incubations [50].
- Quench Endogenous Activity: When using HRP-based detection, quench endogenous peroxidase activity with 3% H₂O₂. For alkaline phosphatase (AP) systems, use 2 mM Levamisole to inhibit endogenous phosphatase [50] [52].
FAQ 4: What is the best method to recover antigenicity while preserving tissue morphology after fixation?
Over-fixation, particularly with cross-linking agents like formalin, can mask epitopes, leading to weak or false-negative staining.
- Problem Identification: Fixation-induced cross-links can hide the target epitope, preventing antibody binding [54] [50].
- Recommended Solutions:
- Heat-Induced Epitope Retrieval (HIER): This is the preferred method for most targets. Heat the sample in a buffer (e.g., citrate buffer at pH 6.0 or Tris/EDTA at pH 9.0) using a microwave oven or pressure cooker. The microwave method is often optimal for restoring immunoreactivity [50].
- Protease-Induced Epitope Retrieval (PIER): For some specific antigens, use enzymes like Proteinase K, Trypsin, or Pepsin. However, PIER requires strict control of incubation time and concentration to avoid destroying tissue morphology and the antigens themselves [54].
Experimental Protocols for Key Methodologies
Protocol 1: Eliminating Non-Specific Adsorption in Molecularly Imprinted Polymers (MIPs) via Surfactant Modification
This protocol details the use of surfactants to suppress non-specific binding on MIPs, as described for the detection of sulfamethoxazole (SMX) [7].
Materials:
- Synthesized MIP and Non-Imprinted Polymer (NIP)
- Surfactant Solution: SDS or CTAB in deionized water
- Target Analyte Solution (e.g., SMX)
- Appropriate buffer (e.g., phosphate buffer)
Procedure:
- Polymer Preparation: Synthesize MIP and NIP using your standard protocol (e.g., bulk or precipitation polymerization with the target molecule as a template).
- Surfactant Treatment: Incubate the polymers with a solution of anionic (SDS) or cationic (CTAB) surfactant. The choice depends on the polymer's external functional groups.
- Equilibration and Binding Test:
- Place the surfactant-modified MIP (and NIP as a control) in vials.
- Add varying concentrations of the target analyte.
- Shake the mixture to reach adsorption equilibrium.
- Separate the polymer and analyze the supernatant to determine the amount of unbound analyte.
- Data Analysis: Calculate the adsorption capacity. Effective modification is indicated by the MIP showing significantly higher adsorption than the NIP, confirming the reduction of non-specific sites.
Validation: The method achieved a limit of detection (LOD) of 6 ng mL−1 for SMX in milk and water samples, with promising recovery rates [7].
Protocol 2: Passivating Porous Silicon (PSi) Biosensors with Zwitterionic Peptides
This protocol describes a novel surface passivation technique to enhance biosensor performance in complex biofluids [51].
Materials:
- PSi thin films
- Zwitterionic peptide (e.g., EKEKEKEKEKGGC)
- Standard coupling reagents: e.g., (3-Aminopropyl)triethoxysilane (APTES), cross-linkers like glutaraldehyde
- Blocking buffer (e.g., 1% BSA)
- Complex test fluids: e.g., GI fluid, serum, bacterial lysate
Procedure:
- Surface Activation: Clean and activate the PSi surface (e.g., via thermal hydrosilylation or APTES/glutaraldehyde chemistry) to generate reactive groups (e.g., amines).
- Peptide Conjugation: Covalently immobilize the zwitterionic peptide onto the activated surface via its terminal cysteine residue. The peptide should be oriented with its charged motifs facing the solution.
- Blocking: After peptide attachment, block any remaining reactive sites with a low concentration of BSA or ethanolamine.
- Aptamer Immobilization (for aptasensors): Immobilize the specific capture aptamer (e.g., for lactoferrin) onto the passivated surface.
- Fouling Test: Expose the sensor to complex biofluids (e.g., 10% serum, GI fluid) and measure the non-specific adsorption signal. Compare it to a control sensor passivated with PEG or BSA.
Validation: Sensors modified with the EK peptide showed more than a tenfold improvement in the limit of detection (LOD) and signal-to-noise ratio over PEG-passivated sensors when detecting lactoferrin in GI fluid [51].
Comparative Data of Antifouling Strategies
Table 1: Performance Comparison of Antifouling Coatings for Biosensors
| Coating Strategy | Material Type | Key Advantage | Tested Sample | Reported Improvement/Performance |
|---|---|---|---|---|
| Zwitterionic Peptide [51] | EKEKEKEKEKGGC | Superior antibiofouling vs. PEG; stable hydration layer | GI fluid, Bacterial lysate | >10x improvement in LOD and signal-to-noise ratio |
| Surfactant Modification [7] | SDS / CTAB | Eliminates non-specific adsorption on MIPs | Milk, Water | LOD for SMX: 6 ng mL⁻¹ |
| Polyethylene Glycol (PEG) [51] | Polymer (750 Da) | Traditional "gold standard" | Various biofluids | Prone to oxidative degradation; lower performance vs. zwitterionic peptides |
| Cross-linked Protein Films [2] | Protein-based | Tunable conductivity and thickness | Serum, Milk | Effective for electrochemical biosensors |
Research Reagent Solutions
Table 2: Essential Reagents for Blocking and Surface Passivation
| Reagent / Material | Function / Application | Key Consideration |
|---|---|---|
| Zwitterionic Peptides [51] | Forms a strong, neutral hydration layer on biosensor surfaces to resist non-specific adsorption of proteins and cells. | Sequence and length can be tuned. EK-based peptides show superior performance. |
| Normal Serums [54] [50] | Used for blocking; contains antibodies that bind to non-specific sites. | Must be from the same species as the secondary antibody. |
| Bovine Serum Albumin (BSA) [54] [52] | A common protein-based blocking agent that binds to non-specific sites on membranes and tissues. | Effective for many applications, but may not be sufficient for highly complex samples. |
| SDS & CTAB [7] | Ionic surfactants used to modify the external surface of Molecularly Imprinted Polymers (MIPs) to eliminate non-specific binding. | Choice depends on the charge of the polymer's external functional groups. |
| Polyethylene Glycol (PEG) [51] | Traditional polymer for surface passivation, forming a hydrophilic, steric barrier. | Susceptible to oxidative degradation in biological media over time. |
| Pre-adsorbed Secondary Antibodies [48] | Secondary antibodies purified to remove cross-reactivity to immunoglobulins of other species. | Critical for multi-species immunofluorescence to prevent cross-talk. |
| Hydrogen Peroxide (H₂O₂) [50] [52] | Used to quench endogenous peroxidase activity in tissues, reducing background in HRP-based detection. | Typically used as a 3% solution. |
Workflow Visualization
Sequential Immunofluorescence (seqIF) Workflow
Surface Passivation Strategy Selection Logic
Evaluating Success: Validation and Comparative Analysis of Antifouling Strategies
FAQ: Troubleshooting Guide for Non-Specific Adsorption
Q1: My immunoassay with serum samples has a high background. What quantitative metrics can I use to diagnose the issue? You can use the Signal-to-Noise Ratio (SNR) and the level of non-specific adsorption to diagnose your assay. A low SNR indicates that the specific signal is being drowned out by background. In one study, a surface coating called Afficoat reduced non-specific adsorption from bovine serum (76 mg/mL protein) to below 0.3 ng/mm², whereas other common coatings like PEG allowed approximately 2.5 ng/mm², demonstrating an over 8-fold improvement in blocking efficiency [3].
Q2: What are the main causes of non-specific adsorption in complex samples like serum? The two primary causes are:
- Hydrophobic Interactions: The driving force comes from the ordered arrangement of water molecules, which leads to a reduction in entropy [55].
- Electrostatic Fields: Surfaces with a charge can attract proteins via the "like repulsion, opposite attraction" principle, depending on the protein's isoelectric point and the system's pH [55]. Serum can contain 40-80 mg/mL of non-specific proteins, which greatly exacerbates this issue [3].
Q3: My negative controls are showing positive signals in my western blot. How can I confirm my antibody's specificity? Perform an immunizing peptide blocking experiment. Pre-incubate your primary antibody with a five-fold excess (by weight) of its specific immunizing peptide. Then, use this "blocked" antibody on a sample identical to the one used with the normal antibody. Any staining that disappears with the blocked antibody is specific to your target [56]. The disappearance of a band or signal provides a qualitative metric for specificity.
Q4: Are there high-throughput methods to quantify antibody responses that minimize non-specific binding issues? Yes, the Biolayer Interferometry Immunosorbent Assay (BLI-ISA) is emerging as a high-throughput alternative to ELISA. It provides quantitative data (binding shift in nm) that correlates with ELISA endpoint titers but reduces manual labor and incubation time, thereby potentially reducing windows for non-specific adsorption. This method is useful for vaccine studies and other applications requiring the quantification of antigen-specific antibodies in sera [57].
Experimental Protocols for Assessment
Protocol 1: Quantifying Non-Specific Adsorption on Sensor Surfaces using SPR
This protocol is adapted from performance tests of the Afficoat surface coating [3].
- Surface Preparation: Functionalize a gold sensor chip with your chosen blocking agent or surface chemistry (e.g., a zwitterionic peptide self-assembled monolayer).
- Baseline Stabilization: Use a phosphate-buffered saline (PBS) buffer to establish a stable baseline in the Surface Plasmon Resonance (SPR) instrument.
- Sample Exposure: Expose the sensor surface to the complex biological sample (e.g., crude bovine serum with a protein concentration of 76 mg/mL) for 20 minutes.
- Rinse: Rinse the sensor surface with PBS for 5 minutes to remove unbound proteins.
- Quantification: The amount of non-specifically adsorbed protein is quantified by the change in the SPR signal (response units), which can be converted to mass per unit area (ng/mm²) [3].
Protocol 2: Validating Antibody Specificity via Immunizing Peptide Blocking
This protocol is used for techniques like western blot and immunohistochemistry [56].
- Preparation: Determine the optimal concentration of your primary antibody for your assay.
- Antibody Dilution: Dilute the antibody to the required volume for two identical experiments and split it into two tubes.
- Blocking: To the first tube ("blocked"), add a five-fold excess (by weight) of the immunizing peptide. To the second tube ("control"), add an equivalent volume of buffer.
- Incubation: Incubate both tubes with agitation at room temperature for 30 minutes or overnight at 4°C.
- Application: Apply the "control" antibody to one sample and the "blocked" antibody to an identical sample.
- Analysis: Perform your staining protocol. Compare the results. The specific signal will be absent or significantly reduced in the sample stained with the "blocked" antibody [56].
The following table summarizes quantitative data on the performance of different surface coatings and assay methods for reducing non-specific adsorption.
Table 1: Performance Metrics of Blocking Strategies and Assays
| Method / Reagent | Key Metric | Performance Result | Application Context |
|---|---|---|---|
| Afficoat (Zwitterionic Peptide SAM) [3] | Non-specific adsorption | < 0.3 ng/mm² after exposure to 76 mg/mL bovine serum | SPR biosensing in crude serum, cell lysate |
| Polyethylene Glycol (PEG) [3] | Non-specific adsorption | ~ 2.5 ng/mm² after exposure to 76 mg/mL bovine serum | Common surface coating for biosensors |
| CM-Dextran [3] | Non-specific adsorption | ~ 4.0 ng/mm² after exposure to 76 mg/mL bovine serum | Common surface coating for biosensors |
| Quantum Dot-Labeled Microplate Immunoassay (QL-MI) [58] | Coefficient of Variation (CV) | Intra-assay: 2.27%; Inter-assay: 8.52% | High-sensitivity CRP detection in clinical serum samples |
| Quantum Dot-Labeled Microplate Immunoassay (QL-MI) [58] | Analytical Recovery | 96.7% - 104.2% | High-sensitivity CRP detection in clinical serum samples |
Research Reagent Solutions
Table 2: Essential Reagents for Reducing Non-Specific Adsorption
| Item | Function | Example Application |
|---|---|---|
| Zwitterionic Peptide Coating (e.g., Afficoat) | Forms a self-assembled monolayer (SAM) that minimizes protein adsorption via hydrophilicity and charge balance [3]. | SPR sensor chips for analyzing biomarkers directly in serum [3]. |
| Blocking Peptides | Specifically binds to and neutralizes a primary antibody's paratope, serving as a negative control to validate specificity [56]. | Immunizing peptide blocking experiments in western blot or IHC [56]. |
| Bovine Serum Albumin (BSA) | A classic blocking protein used to cover residual binding sites on surfaces (e.g., microplates, membranes) [56] [58]. | Used in blocking buffers for ELISA and western blot [56]. |
| Monoclonal Antibodies (mAbs) to Distinct Epitopes | Enhance assay specificity by forming a sandwich complex that minimizes cross-reactivity in immunoassays [58]. | Quantum dot-based microplate immunoassays for CRP [58]. |
Workflow and Relationship Diagrams
Troubleshooting Non-Specific Adsorption
Workflow for Surface Preparation
Comparative Performance of Different Surface Chemistries and Coatings
Frequently Asked Questions (FAQs)
1. What is non-specific adsorption (NSA) and why is it a problem in biosensing? Non-specific adsorption (NSA), also known as non-specific binding or biofouling, occurs when molecules from a complex sample (like serum) adhere indiscriminately to your sensor's surface through physisorption [1]. This is driven by hydrophobic interactions, ionic interactions, van der Waals forces, and hydrogen bonding [1] [2]. In serum samples, which can contain 40-80 mg/mL of protein, NSA leads to elevated background signals, false positives, reduced sensitivity and specificity, and unreliable data by masking the signal from your target analyte [1] [3].
2. For analyzing targets in serum, should I choose a PEG-based coating or a zwitterionic peptide coating? For the most challenging serum samples, zwitterionic peptide coatings like Afficoat have demonstrated superior performance. A comparative study exposed different coatings to bovine serum (76 mg/mL protein) and measured the resulting non-specific adsorption [3]. The results, summarized in the table below, show that the zwitterionic peptide SAM (Afficoat) outperformed others. While PEG is a good option, zwitterionic peptides currently represent the state-of-the-art for minimizing fouling from concentrated serum [3].
Table: Comparison of Non-Specific Adsorption Levels for Different Surface Coatings in Bovine Serum
| Surface Coating | Relative Non-Specific Adsorption Level | Key Characteristics |
|---|---|---|
| Zwitterionic Peptide SAM (Afficoat) | Lowest | Hydrophilic, zwitterionic; forms a self-assembled monolayer on gold [3]. |
| PEG | Medium | Well-established; hydrophilic polymer that resists protein adsorption [3]. |
| CM-Dextran | Highest | Hydrogel network; commonly used in SPR but prone to higher fouling in complex samples [3]. |
3. My sensor surface is already fouled after testing a serum sample. Can I clean it and reuse it? This depends heavily on the surface chemistry and the strength of the adsorbed layers. For covalently stable coatings like cross-linked polymers or SAMs, rigorous cleaning regimens using surfactant solutions (e.g., SDS) or acidic/basic washes can sometimes regenerate the surface [1] [4]. However, repeated cleaning can degrade delicate coatings. For disposable sensor strips or chips where surface integrity is critical for quantitative accuracy, single use is strongly recommended to avoid carryover and inconsistent performance [59].
4. Besides surface coatings, what other experimental parameters can I adjust to minimize NSA from serum? Yes, optimizing your running buffer is a critical and simple first step. Several additives can help shield your analyte and surface from non-specific interactions [60]:
- Additives: Incorporate bovine serum albumin (BSA) at ~1% or non-ionic surfactants like Tween 20 at low concentrations (e.g., 0.05%) to block hydrophobic surfaces and prevent analyte loss to tubing [60].
- pH Adjustment: Adjust the buffer pH to the isoelectric point (pI) of your analyte or away from the pI of common foulants to neutralize charge-based attractions [60].
- Salt Concentration: Increase the ionic strength (e.g., with 150-200 mM NaCl) to shield electrostatic interactions between charged proteins and the sensor surface [60].
Troubleshooting Guide
Problem: High Background Signal in Serum Samples
Possible Cause 1: Inadequate or sub-optimal surface coating. The chosen surface chemistry is not sufficiently resistant to the high concentration of proteins and other biomolecules present in serum.
Solution:
- Switch to a more robust coating: Move from traditional coatings like dextran to ultralow-fouling options such as zwitterionic peptides (e.g., Afficoat) or high-density PEG [6] [3].
- Employ a combined approach: Use a dedicated low-fouling coating in conjunction with buffer additives like BSA or Tween 20 for layered protection [60].
Possible Cause 2: Electrostatic interactions between serum proteins and the sensor surface. The surface may have a net charge that attracts oppositely charged proteins in the serum.
Solution:
- Adjust buffer pH: Modify the pH of your running buffer and sample diluent to ensure both the analyte and the surface coating are at a neutral or repulsive charge state [60].
- Increase ionic strength: Add salt (e.g., NaCl) to your buffer to shield electrostatic charges. A concentration of 200 mM NaCl has been shown to effectively reduce NSB of charged analytes like IgG [60].
Possible Cause 3: Hydrophobic interactions. Hydrophobic patches on the sensor surface or on proteins promote irreversible adsorption.
Solution:
- Add a non-ionic surfactant: Include a mild detergent like Tween 20 (0.01-0.1%) in your buffers to disrupt hydrophobic interactions [60] [4].
- Ensure surface hydrophilicity: Select coatings that create a well-hydrated, hydrophilic boundary layer, which is a key feature of PEG and zwitterionic materials [1] [61].
Problem: Low Specific Signal or False Negatives in Serum
Possible Cause: The antifouling coating is interfering with the biorecognition element. The passivation layer might be too thick or dense, causing steric hindrance that prevents the target analyte from accessing the captured probe (e.g., antibody).
Solution:
- Optimize coating density: If possible, tune the density of your antifouling layer (e.g., use a mixed SAM) to balance fouling resistance with probe accessibility [1] [7].
- Use a capture system: Immobilize your bioreceptor using a capture system, such as the high-affinity biotin-streptavidin system, which can help present the receptor more uniformly and accessibly above the antifouling layer [59].
Problem: Inconsistent Results Between Different Serum Samples
Possible Cause: Variable composition of individual serum samples. Different donors or patient sera can have varying levels of lipids, immunoglobulins, and other components, leading to inconsistent NSB that is hard to control with a single blank subtraction [62].
Solution:
- Implement a on-chip reference subtraction: Use a sophisticated blank correction method. As demonstrated for anti-HLA antibody detection, capture a "non-cognate target" (a structurally similar but irrelevant protein) on the same flow cell in a separate cycle and subtract its signal from that of your specific target. This accounts for sample-specific NSB more accurately than a generic blank [62].
Key Experimental Protocols
Protocol 1: Evaluating Coating Performance with Surface Plasmon Resonance (SPR)
This protocol is adapted from studies that tested various self-assembled monolayers (SAMs) against crude serum [3].
1. Objective: To quantitatively compare the non-specific adsorption resistance of different surface chemistries when exposed to complex biological serum. 2. Materials: * SPR instrument with a gold sensor chip. * Coating reagents (e.g., PEG-thiol, zwitterionic peptide-thiol). * Phosphate Buffered Saline (PBS), pH 7.4. * Crude bovine or human serum (un-diluted or minimally diluted). 3. Procedure: * Surface Preparation: Immobilize the different coatings (e.g., Candidate A, Candidate B) onto separate flow channels of a gold sensor chip according to their specific protocols (e.g., incubating with thiol solutions to form SAMs). * Baseline Stabilization: Prime the SPR system with PBS until a stable baseline is achieved. * Serum Exposure: Expose all coated surfaces to the crude serum sample for 20 minutes at a constant flow rate. * Rinse: Rinse the system with PBS for 5 minutes to remove loosely bound material. * Data Analysis: Measure the change in resonance units (ΔRU) after the rinse step. A lower ΔRU indicates a superior coating with higher resistance to non-specific adsorption.
The workflow for this evaluation is outlined below.
Protocol 2: Reducing NSA in SPR via Buffer Optimization and Reference Subtraction
This protocol is ideal for researchers who need to work with existing sensor chips and must mitigate NSA through solution-based methods and data processing [60] [62].
1. Objective: To suppress NSA in serum-containing samples through buffer additives and a robust blank subtraction strategy. 2. Materials: * SPR instrument and sensor chip with immobilized ligand. * Running buffer (e.g., HBS-EP). * Buffer additives: BSA, Tween 20, NaCl. * Serum sample. * Non-cognate target protein (for advanced subtraction). 3. Procedure: * Buffer Screening: Prepare running buffers containing different additives: * Condition 1: Base buffer + 0.05% Tween 20. * Condition 2: Base buffer + 1% BSA. * Condition 3: Base buffer + 200 mM NaCl. * Preliminary Test: Inject your serum sample over a bare or non-specific surface and measure NSB in each buffer condition to identify the most effective one. * Ligand Immobilization: Immobilize your specific target ligand on the sensor chip. * Reference Channel Setup: If possible, use a reference channel immobilized with a non-cognate but structurally similar protein [62]. * Sample Injection: Run your serum sample in the optimized buffer. * Data Processing: Subtract the signal from the reference channel (or a blank run without serum) from the ligand channel to isolate the specific binding signal.
The Scientist's Toolkit: Essential Reagents & Materials
Table: Key Research Reagent Solutions for Combating Non-Specific Adsorption
| Item Name | Function/Brief Explanation | Example Use Case |
|---|---|---|
| Zwitterionic Peptide SAM (Afficoat) | Forms an ultralow-fouling monolayer; hydrophilic and zwitterionic properties create a strong hydration layer that resists protein adsorption [3]. | Gold sensor chip functionalization for direct analysis in serum and plasma [3]. |
| Polyethylene Glycol (PEG) | A hydrophilic polymer that sterically hinders the approach of proteins to the surface, reducing fouling [1]. | A common passive coating for biosensors and nanoparticles to improve biocompatibility [1] [61]. |
| Bovine Serum Albumin (BSA) | A "blocker" protein that adsorbs to vacant sites on the surface, preventing subsequent non-specific adsorption of other proteins [60]. | Added to buffers (typically 1%) or used as a pre-incubation step to block unused binding sites on surfaces [60]. |
| Tween 20 | A non-ionic surfactant that disrupts hydrophobic interactions, a major driver of NSA [60] [4]. | Added to running and sample buffers at low concentrations (0.01-0.1%) to minimize hydrophobic binding [60]. |
| Sodium Chloride (NaCl) | Increases the ionic strength of the solution, producing a shielding effect that reduces charge-based (electrostatic) interactions [60]. | Added to buffers (e.g., 150-200 mM) to minimize NSA of charged analytes like IgG [60]. |
| Low-Adsorption Consumables | Tubes and plates with surface treatments (e.g., polymer coatings) that minimize binding of precious or sticky samples like proteins and nucleic acids [4]. | Sample collection and storage for sensitive analytes prone to loss via adsorption to container walls [4]. |
For researchers, scientists, and drug development professionals, achieving reliable assay results in complex media like whole serum and cell lysates is a significant hurdle. These samples present a challenging environment characterized by a high concentration of diverse proteins, lipids, and other biomolecules that can interfere with detection. A primary source of this interference is non-specific adsorption (NSA)—the unwanted adhesion of non-target molecules to sensor surfaces or assay components [1] [2]. NSA leads to elevated background signals, false positives, reduced sensitivity, and poor reproducibility, ultimately compromising data validity [1] [63]. This guide provides targeted troubleshooting and methodologies to help you validate your assays effectively in these complex environments.
Troubleshooting FAQs on NSA in Complex Media
1. Our immunoassays in serum samples consistently show high background signal. What are the primary strategies to reduce this non-specific adsorption?
High background in complex samples like serum is often due to NSA from abundant proteins (e.g., albumin, immunoglobulins) and other components. Your strategy should be multi-layered, focusing on surface preparation, blocking, and careful reagent selection.
- Optimize Your Blocking Solution: The choice of blocker is critical. Do not assume one blocker works for all assays. Test different blockers side-by-side to find the most effective one for your specific sample matrix.
- Common Blockers: Bovine Serum Albumin (BSA), casein, non-fat dry milk (NFDM), fish gelatin, and polyethylene glycol (PEG) [1] [39].
- Considerations: Be aware that cross-reaction can occur; for example, some secondary antibodies may recognize proteins in NFDM or BSA [39]. In such cases, use a blocker that does not contain the interfering protein.
- Select the Appropriate Assay Surface: The binding capacity of your microplate can influence NSA.
- Guidance: Use high-binding plates for capturing low-abundance targets but be vigilant about potential NSA. Medium-binding plates may offer a better balance for complex samples by reducing the surface area available for non-specific interactions [39].
- Employ Advanced Surface Coatings: Move beyond simple protein blockers by using engineered non-fouling materials.
- Leverage Additives and Novel Strategies: Incorporate reagents into your sample or assay buffer that dynamically suppress NSA.
- Detergents: Add low concentrations of detergents like Tween-20 to your wash buffers.
- Amphiphilic Sugars: Recent studies show that additives like
n-Dodecyl β-D-maltosidecan reversibly block hydrophobic surfaces, reducing NSA when added directly to the analyte solution [15]. - Dual-Layer Membrane Cloaking (DLMC): This innovative method involves forming a temporary, removable membrane (e.g., didodecyldimethylammonium bromide, DDAB) with an additional BSA layer over your immunosensor. After incubation with the sample, this dual-layer is stripped away, selectively removing non-specifically adsorbed proteins while leaving specific complexes intact [64].
2. How can we validate that our detection antibody is specific and not cross-reacting with other proteins in a cell lysate?
Antibody cross-reactivity is a major source of false positives and must be rigorously addressed during assay development [65] [39].
- Use Genetically Validated Controls: The most robust method is to use knockout (KO) cell lines or lysates. When your target protein is absent, any signal generated indicates cross-reactivity or non-specific binding [65].
- Test Antibody Specificity with Lysate Panels: Perform western blot or ELISA on a panel of cell lysates or tissue homogenates from different sources. A specific antibody should produce a signal only in samples known to express the target and should recognize a single band at the expected molecular weight in western blots (though post-translational modifications can cause multiple bands) [65].
- Validate with an Orthogonal Method: Confirm your ELISA or biosensor results using a different, non-antibody-based technique, such as mass spectrometry (MS), if possible [65].
- Check for Cross-Reaction of Secondary Reagents: In indirect or sandwich ELISA formats, ensure your enzyme-labeled secondary antibodies or streptavidin conjugates do not bind directly to your capture antibody or other sample proteins.
- Experimental Check: Run a control well with your capture antibody and detection system, but omit the primary/detection antibody. A signal in this well indicates direct binding of your secondary reagent to the capture antibody, necessitating the use of cross-adsorbed secondary antibodies or antibodies from different host species [39].
3. What are the best practices for directly analyzing serum or lysates without sample dilution, while maintaining sensitivity?
Direct analysis minimizes sample preparation but requires robust anti-fouling strategies.
- Implement a "Membrane Cloaking" Strategy: As mentioned in the DLMC method, this approach is specifically designed for direct analysis of serum. It allows you to incubate the complex sample directly on the sensor, followed by the removal of the cloaking layer and the adsorbed interferents, enabling ultra-sensitive detection without dilution [64].
- Utilize Affinity Capture on Non-Fouling Substrates: For techniques like cryo-EM or other capture assays, modify surfaces with antifouling agents. For example, graphene oxide (GO) sheets functionalized with NTA for His-tagged protein capture can be further treated with BSA to block non-specific binding from cell lysates, allowing selective target isolation [66].
- Focus on Highly Sensitive Biosensor Platforms: Leverage the intrinsic advantages of microfluidic biosensors, which use small sample volumes and can enhance the signal-to-noise ratio by reducing background signals [1]. Coupled with the appropriate surface chemistry, these can be powerful for direct analysis.
Essential Experimental Protocols
Protocol 1: Evaluating Blocking Agents for ELISA in Serum-Containing Samples
This protocol helps you systematically identify the optimal blocking agent to minimize NSA in your ELISA when working with serum.
Key Materials:
- Microplates: High-binding and medium-binding 96-well polystyrene plates [39].
- Blocking Agents: Prepare solutions of BSA, casein, non-fat dry milk (NFDM), fish gelatin, etc. [39].
- Sample Model: Diluted serum spiked with your target antigen.
- Detection System: Validated primary and secondary antibodies.
Methodology:
- Coat the plate with your capture antibody or antigen as per your standard protocol.
- Blocking: Divide the plate and add different blocking solutions to separate wells. Incubate for 1-2 hours at room temperature.
- Wash the plate thoroughly.
- Add Sample: Add your serum-spiked sample and your positive/negative controls to the wells.
- Proceed with Detection: Complete your standard ELISA protocol (detection antibody, enzyme conjugate, substrate).
- Data Analysis: Calculate the signal-to-noise ratio (Signal from positive control / Signal from negative control) for each blocker. The blocker yielding the highest ratio is likely the most effective.
Protocol 2: Direct Chronoamperometric Immunoassay of Serum Using Dual-Layer Membrane Cloaking (DLMC)
This detailed protocol, adapted from biosensor research, enables direct and sensitive detection in serum without dilution or separation [64].
Key Materials:
- Sensor Substrate: e.g., ITO chip or SPR sensor chip.
- Chemicals: Didodecyldimethylammonium bromide (DDAB), Bovine Serum Albumin (BSA), Triton X-100 (5% v/v).
- Antibodies: Target-specific capture antibody (e.g., Mouse IgG - MIgG).
- Solution: 3-mercaptopropionic acid (MPA), EDAC/NHS coupling reagents.
Methodology:
- Sensor Functionalization: Immobilize the capture antibody (MIgG) on the sensor substrate. For example, form a self-assembled monolayer (SAM) of MPA on a gold surface, activate with EDAC/NHS, and couple MIgG [64].
- Form First Cloaking Layer: Fuse a DDAB membrane onto the MIgG-modified surface.
- Form Second Cloaking Layer: Adsorb a thin layer of BSA onto the DDAB membrane to create the BSA/DDAB Dual-Layer Membrane (DLM). This layer resists penetration of matrix proteins.
- Sample Incubation: Incubate the sensor with the raw serum sample containing the analyte. Specific binding occurs between the analyte and the capture antibody, while non-specific proteins adsorb to the removable DLM.
- Membrane and NSA Removal: Remove the BSA/DDAB DLM by washing with 5% Triton X-100. This step selectively shears away the cloaking layer and all non-specifically adsorbed proteins.
- Final Measurement: Perform the final detection step (e.g., chronoamperometry with an enzyme conjugate) on the cleaned sensor surface. The specific antibody-analyte complexes remain undisturbed [64].
Data Presentation
Table 1: Performance Comparison of Common Blocking Agents in ELISA This table summarizes data from systematic evaluations of blockers, highlighting their performance in reducing non-specific binding [39].
| Blocking Agent | Optimal Concentration | Relative Background Signal | Key Considerations |
|---|---|---|---|
| BSA | 1-5% | Medium | High purity required; potential for cross-reaction with anti-BSA antibodies. |
| Casein | 1-3% | Low | Excellent blocker; may not be suitable for phospho-specific antibodies. |
| Non-Fat Dry Milk | 1-5% | Low | Very effective but contains casein and whey; high potential for cross-reactivity. |
| Fish Gelatin | 1-5% | Medium-High | Good alternative to mammalian protein blockers for reducing cross-species reactivity. |
| PVP/PEG | 0.1-1% | Variable | Synthetic polymers; require concentration optimization for each assay. |
Table 2: Cross-Reaction Check for Secondary Antibodies in Sandwich ELISA This control experiment is critical for validating assay specificity. A signal in the "No Detector Ab" control indicates cross-reaction [39].
| Well Contents | Expected Result | Interpretation of a Signal |
|---|---|---|
| Capture Ab + Target + Detector Ab + Secondary Ab | Signal | Valid specific binding. |
| Capture Ab + Target + Secondary Ab (No Detector Ab) | No Signal | Signal indicates secondary Ab binds directly to Capture Ab. |
| Capture Ab + Serum + Detector Ab + Secondary Ab | Signal | Valid specific binding in complex media. |
| Capture Ab + Serum + Secondary Ab (No Detector Ab) | No Signal | Signal indicates secondary Ab binds to serum proteins. |
Experimental Workflow Visualization
The following diagram outlines a logical workflow for developing and validating an assay for use in complex media, integrating the key concepts from this guide.
The Scientist's Toolkit: Key Research Reagent Solutions
Table 3: Essential Reagents for Combating NSA This table lists key reagents, their functions, and application contexts to assist in experimental planning.
| Reagent | Function / Purpose | Example Application Context |
|---|---|---|
| Zwitterionic Polymers (e.g., pSBMA) | Forms a highly hydrated, non-fouling surface that resists protein adsorption. | Coating for SPR biosensors and electrochemical sensors for direct serum analysis [64] [2]. |
| Amphiphilic Sugars (e.g., n-Dodecyl β-D-maltoside) | Acts as a reversible blocking agent by adsorbing to hydrophobic surfaces; added to analyte solutions. | Reducing NSA in label-free immunoassays with simple surface chemistry [15]. |
| Didodecyldimethylammonium Bromide (DDAB) | Forms a supported bilayer membrane (SBM) for use as a "cloaking" layer. | Key component of the Dual-Layer Membrane Cloaking (DLMC) method for direct serum analysis [64]. |
| Cross-Adsorbed Secondary Antibodies | Purified to remove antibodies that bind to immunoglobulins from non-target species. | Critical for sandwich ELISAs to prevent cross-reaction between the detection system and the capture antibody [39]. |
| His-Tag Purification Systems (GO-NTA grids) | Graphene oxide functionalized with NTA for capturing His-tagged proteins; can be treated with BSA to reduce background. | Selective capture of target proteins directly from raw cell lysates for structural biology (cryo-EM) [66]. |
Conclusion
Effectively mitigating non-specific adsorption in serum samples is not a one-size-fits-all endeavor but requires a multifaceted strategy that integrates sample preparation, sophisticated surface chemistry, and rigorous validation. The journey begins with a solid understanding of the fundamental interactions between serum proteins and sensor surfaces, progresses through the careful application and optimization of blocking agents and antifouling materials, and is validated with robust analytical techniques. Promising future directions include the development of tunable, multi-functional coatings, the integration of high-throughput screening for new materials, and the application of machine learning to predict and design superior antifouling surfaces. By systematically addressing NSA, researchers can significantly enhance the accuracy and translational potential of their biosensors and immunoassays, paving the way for more reliable diagnostics and biomedical research outcomes.
References
- 1. Non-Specific Adsorption Reduction Methods in Biosensing [https://pmc.ncbi.nlm.nih.gov/articles/PMC6603772/]
- 2. Promising Solutions to Address the Non-Specific ... [https://www.mdpi.com/2227-9040/13/3/92]
- 3. Afficoat™ for Reducing Non-Specific Adsorption in SPR ... [https://www.affiniteinstruments.com/post/afficoat-for-reducing-non-specific-adsorption]
- 4. Nonspecific Binding: Main Factors of Occurrence and ... [https://dmpkservice.wuxiapptec.com/articles/44-nonspecific-binding-main-factors-of-occurrence-and-strategies/]
- 5. Managing nonspecific adsorption to liquid chromatography ... [https://www.sciencedirect.com/science/article/abs/pii/S0003267023002155]
- 6. Non-specific adsorption of serum and cell lysate on 3D ... [https://www.sciencedirect.com/science/article/pii/S1878535215002099]
- 7. Innovative approaches to suppress non-specific adsorption ... [https://www.sciencedirect.com/science/article/abs/pii/S0956566324000563]
- 8. Mitigation of Non-Specific Adsorption on HPLC Systems ... [https://www.waters.com/nextgen/us/en/library/application-notes/2023/mitigation-of-non-specific-adsorption-on-hplc-systems-using-maxPeak-premier-columns.html?srsltid=AfmBOoqHf9rZxMmwuL58SdWDfKohMoSowCmMZYnoI3u88ICb0mCDcyAl]
- 9. Optimizing drug discovery: Surface plasmon resonance ... [https://www.sciencedirect.com/science/article/pii/S2667022423002530]
- 10. An Introduction To Immunoassay Interference | Blog [https://www.biosynth.com/blog/immunoassay-interference]
- 11. 2-Stage Factor VIII Assays [https://practical-haemostasis.com/Factor%20Assays/2_stage_factor_assays.html]
- 12. Clinical utility and impact of the use of the chromogenic vs ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6916414/]
- 13. Crosslinking of human plasma C-reactive protein to ... [https://www.sciencedirect.com/science/article/pii/S2213231721000732]
- 14. Non-specific adsorption of protein to microfluidic materials [https://www.sciencedirect.com/science/article/abs/pii/S0927776521005828]
- 15. Reduction of non-specific adsorption in label-free assays ... [https://www.sciencedirect.com/science/article/pii/S0925400522002994]
- 16. What are Some Potential Errors that can Occur in an ELISA? [https://shop.surmodics.com/elisa-test-results-troubleshooting]
- 17. Difficulty in obtaining signal for ELISA [https://www.abcam.com/en-us/technical-resources/troubleshooting/signal-problems-elisa]
- 18. How to Minimize False Positives in ADP Detection [https://bellbrooklabs.com/how-to-minimize-false-positives-in-adp-detection/?srsltid=AfmBOoqQwPFUTjsanOO7dDwICeznKFJGVqzpd9-QPuuhvmEXGpbspKTM]
- 19. Interference with Fluorescence and Absorbance - NCBI - NIH [https://www.ncbi.nlm.nih.gov/books/NBK343429/]
- 20. Abundant Protein Depletion for Mass Spectrometry [https://www.thermofisher.com/us/en/home/life-science/protein-biology/protein-mass-spectrometry-analysis/sample-prep-mass-spectrometry/abundant-protein-depletion-mass-spectrometry.html]
- 21. A simple serum depletion method for proteomics analysis [https://pubmed.ncbi.nlm.nih.gov/32372655/]
- 22. Troubleshooting Tips for Adsorption and Chromatography [https://www.linkedin.com/advice/0/how-do-you-troubleshoot-resolve-common-1c]
- 23. Mistakes and inconsistencies regarding adsorption of ... [https://www.sciencedirect.com/science/article/abs/pii/S0043135417302695]
- 24. Conformational flexibility of fatty acid-free bovine serum ... [https://www.nature.com/articles/s43246-020-0047-9]
- 25. An Improved Surface Passivation Method for Single- ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4245390/]
- 26. Non-specific adsorption: Common causes, how it affects ... [https://www.selectscience.net/article/non-specific-adsorption-common-causes-how-it-affects-chromatographic-results-and-how-to-prevent-it]
- 27. Preparation and Optimization of Bovine Serum Albumin ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10670644/]
- 28. Recent Advances in Antifouling Materials for Surface ... [https://www.mdpi.com/2073-4360/13/12/1929]
- 29. Anti-fouling Coatings of Poly(dimethylsiloxane) Devices for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4414934/]
- 30. Control of Specific/Nonspecific Protein Adsorption [https://pmc.ncbi.nlm.nih.gov/articles/PMC8706203/]
- 31. Effect of Surfactants on the Binding Properties of a Molecularly ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9741244/]
- 32. Small Toxic Molecule Detection and Elimination Using ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12190631/]
- 33. Advances in biomass-based molecularly imprinted polymers [https://pubs.rsc.org/en/content/articlehtml/2025/gc/d5gc01018g]
- 34. Enzyme Linked Immunosorbent Assay - StatPearls - NCBI - NIH [https://www.ncbi.nlm.nih.gov/books/NBK555922/]
- 35. ELISA Troubleshooting Guide: Solutions for Common ... [https://product.atagenix.com/archive/1073.html]
- 36. Nonspecificity fingerprints for clinical-stage antibodies in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10756282/]
- 37. Apparent Activity in High-Throughput Screening: Origins ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC2878863/]
- 38. Blocking Buffers for Western Blot and ELISA [https://www.thermofisher.com/us/en/home/life-science/protein-biology/protein-biology-learning-center/protein-biology-resource-library/pierce-protein-methods/blocking-buffers-western-blot-elisa.html]
- 39. Non-Specific Binding and Cross-Reaction of ELISA [https://pmc.ncbi.nlm.nih.gov/articles/PMC8394222/]
- 40. Blocking Buffer Optimization Protocol [https://www.licorbio.com/support/contents/applications/western-blots/blocking-buffer-optimization-protocol.html]
- 41. Optimization of blocking conditions for fluorescent Western ... [https://pubmed.ncbi.nlm.nih.gov/32014414/]
- 42. Effect of Ionic Strength on Initial Interactions of Escherichia ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC94024/]
- 43. The interactions of non-functionalized polystyrene ... [https://www.nature.com/articles/s41598-025-15422-w]
- 44. Hemocompatible hemoadsorbent for effective removal of ... [https://www.sciencedirect.com/science/article/abs/pii/S0021979719308276]
- 45. Zwitteration: coating surfaces with zwitterionic functionality to reduce ... [https://pubmed.ncbi.nlm.nih.gov/24754399/]
- 46. One-step polymer surface modification for minimizing drug, protein... [https://orbit.dtu.dk/en/publications/one-step-polymer-surface-modification-for-minimizing-drug-protein]
- 47. Self-Assembly Strategy for Reducing - Non on... Specific Adsorption [https://www.researchsquare.com/article/rs-1091250/v1]
- 48. Blocking Cross-Species Secondary Binding When ... [https://www.frontiersin.org/journals/neuroscience/articles/10.3389/fnins.2021.579859/full]
- 49. Multiple Antigen Immunostaining Procedures - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC7670878/]
- 50. Immunohistochemistry (IHC) Troubleshooting Guide & the ... [https://www.cellsignal.com/learn-and-support/troubleshooting/ihc-troubleshooting-guide?srsltid=AfmBOoo_hhal-X_Jq9PIWG5Vet9synAb4EOx0ZcmoiEJmiYIUH0c15ml]
- 51. Porous silicon biosensors meet zwitterionic peptides [https://pubs.rsc.org/en/content/articlehtml/2025/nh/d5nh00478k]
- 52. IHC Troubleshooting Guide | Common Issues & Fixes [https://www.bosterbio.com/protocol-and-troubleshooting/ihc-troubleshooting?srsltid=AfmBOorYsGy13kvUkT5hV2KFTyNy3BQnH-vRfmWyA8_nbH-t-2FpagK3]
- 53. Ten Approaches That Improve Immunostaining: A Review ... [https://www.mdpi.com/1422-0067/23/3/1426]
- 54. An introduction to Performing Immunofluorescence Staining [https://pmc.ncbi.nlm.nih.gov/articles/PMC6918834/]
- 55. How to efficiently suppress non - specific ? - LNJNBIO adsorption [https://www.lingjunbio.com/how-to-efficiently-suppress-non-specific-adsorption/]
- 56. Blocking with immunizing peptide protocol [https://www.abcam.com/en-us/technical-resources/protocols/blocking-with-immunizing-peptides]
- 57. A high-throughput multi-species platform using Biolayer ... [https://www.sciencedirect.com/science/article/pii/S0022175925001802]
- 58. Sensitive and rapid quantification of C-reactive protein using ... [https://translational-medicine.biomedcentral.com/articles/10.1186/1479-5876-10-24]
- 59. Suppressing Non-Specific Binding of Proteins onto ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6468902/]
- 60. 4 Ways to Reduce Non-Specific Binding in SPR Experiments [https://nicoyalife.com/blog/4-ways-reduce-non-specific-binding-spr/]
- 61. Surface modifications of biomaterials in different applied ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10331796/]
- 62. Overcoming non-specific binding to measure the active ... [https://www.sciencedirect.com/science/article/abs/pii/S0956566318304433]
- 63. Validation Processes of Protein Biomarkers in Serum—A ... [https://www.mdpi.com/1424-8220/12/9/12710]
- 64. Investigation of dual-layer membrane cloaking method by ... [https://www.sciencedirect.com/science/article/abs/pii/S0956566311005033]
- 65. Antibody validation for Western blot: By the user, for the user [https://pmc.ncbi.nlm.nih.gov/articles/PMC6983856/]
- 66. Selective Capture of Histidine-tagged Proteins from Cell ... [https://www.nature.com/articles/srep32500]