Surface Plasmon Resonance (SPR) Fundamentals: A Comprehensive Guide for Drug Discovery and Biomedical Research
This article provides a comprehensive overview of Surface Plasmon Resonance (SPR), a powerful, label-free optical technique for real-time biomolecular interaction analysis.
Surface Plasmon Resonance (SPR) Fundamentals: A Comprehensive Guide for Drug Discovery and Biomedical Research
Abstract
This article provides a comprehensive overview of Surface Plasmon Resonance (SPR), a powerful, label-free optical technique for real-time biomolecular interaction analysis. Tailored for researchers, scientists, and drug development professionals, it covers the foundational physics of SPR, detailed methodological protocols for kinetic and affinity studies, advanced troubleshooting and optimization strategies for robust data generation, and validation through contemporary case studies in diagnostics and drug discovery. The content synthesizes core principles with practical applications, highlighting SPR's critical role in accelerating therapeutic development and clinical diagnostics.
Understanding SPR: Core Principles and the Physics of Biomolecular Sensing
What is Surface Plasmon Resonance? Defining the Label-Free Detection Technique
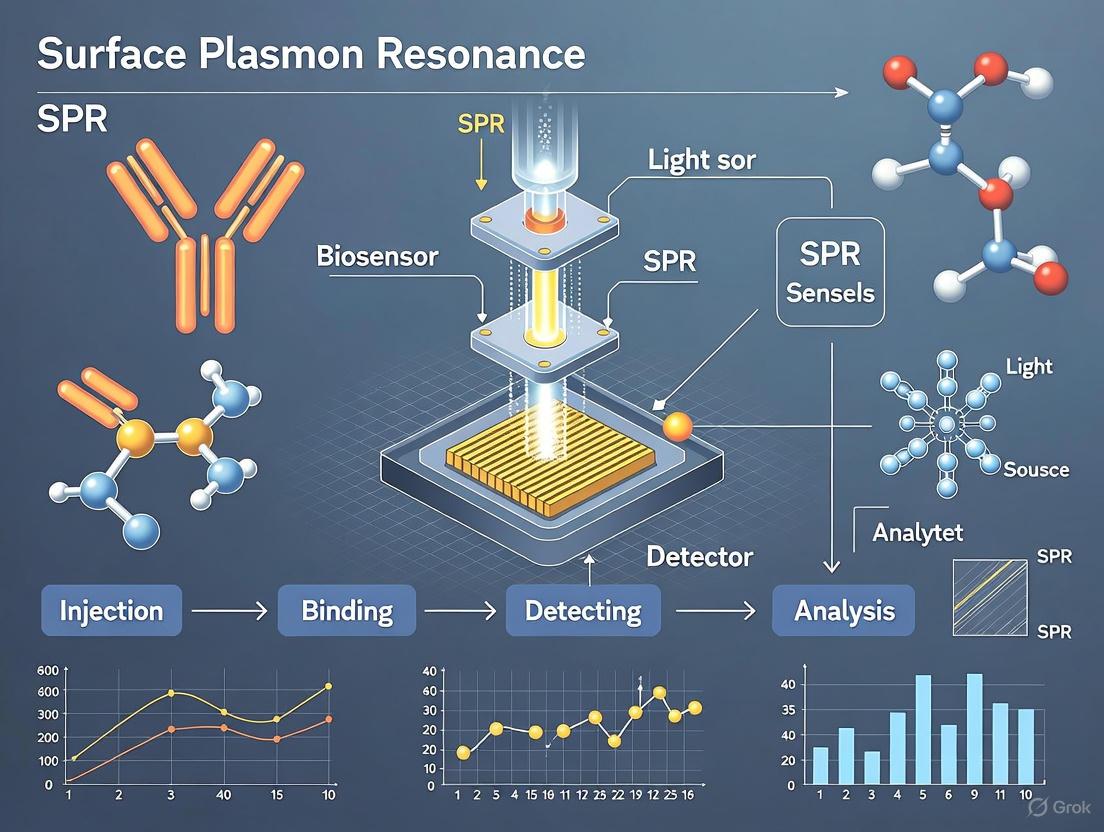
Surface Plasmon Resonance (SPR) is a powerful optical technique that enables the real-time, label-free detection and quantification of molecular interactions by measuring changes in the refractive index on a sensor surface. This phenomenon occurs when plane-polarized light hits a metal film under total internal reflection conditions, exciting collective oscillations of free electrons known as surface plasmons. The core principle underpinning SPR biosensing is that the resonance condition is exquisitely sensitive to changes in the local refractive index, which alters when molecules bind to or dissociate from the sensor surface. First developed for analytical applications in the 1980s, SPR has since become an indispensable tool in biochemical research, drug discovery, and diagnostic development, providing critical insights into binding kinetics, affinity, and specificity without requiring fluorescent or radioactive labels.
Core Principles of Surface Plasmon Resonance
The Physical Phenomenon
At its most fundamental level, Surface Plasmon Resonance exploits the wave-like nature of electrons at a metal-dielectric interface. When conduction electrons in a thin metal film are excited by incident light at a specific angle, they oscillate collectively—a quantum phenomenon known as a surface plasmon polariton (SPP). These SPPs are electromagnetic waves that propagate along the metal surface, with their electric field intensity decaying exponentially perpendicular to the interface, typically penetrating the medium above the surface by about 100 to 200 nanometers [1]. This limited penetration depth makes SPR exceptionally sensitive to minute changes occurring at the sensor surface.
The resonance condition is highly dependent on the refractive index of the medium adjacent to the metal film. When molecules (such as proteins or DNA) bind to the surface, they displace the aqueous buffer medium, increasing the local refractive index. This change shifts the resonance angle—the specific angle of incident light at which maximum energy transfer occurs to the surface plasmons, resulting in a characteristic "dip" in the intensity of reflected light. By monitoring this angle in real time, researchers can precisely track binding events as they happen [2] [1].
Standard Experimental Configuration
The most common implementation of SPR uses the Kretschmann configuration to efficiently excite surface plasmons. In this setup:
- A thin metal film (typically gold or silver, 50-100 nm thick) is coated onto a glass prism or slide.
- P-polarized light (with its electric field component parallel to the plane of incidence) is directed through the prism onto the metal film under conditions of total internal reflection.
- An evanescent wave penetrates the metal film and, at the specific resonance angle, couples with the free electrons to excite surface plasmons.
- A detector measures the intensity of reflected light, identifying the angle of minimum reflectivity (the resonance angle) [2] [3].
This configuration enables the highly sensitive detection of molecular interactions by immobilizing one binding partner (the ligand) on the sensor chip surface and flowing the other partner (the analyte) over it in solution. The binding-induced refractive index changes are measured in resonance units (RU), where 1 RU typically corresponds to a change of 10⁻⁴ degrees in resonance angle and is approximately proportional to 1 pg/mm² of protein mass on the surface [4].
Table 1: Key Components in a Standard SPR Experiment
| Component | Description | Function in SPR |
|---|---|---|
| Sensor Chip | Glass substrate coated with thin gold film and functionalized matrix | Provides surface for ligand immobilization and plasmon excitation |
| Running Buffer | Aqueous buffer, often with detergent (e.g., 0.05% Tween 20) | Maintains constant pH and ionic strength; reduces non-specific binding |
| Ligand | Molecule immobilized on sensor surface (e.g., protein, DNA) | Serves as capture molecule for binding partner |
| Analyte | Molecule in solution flowed over ligand surface | Binding partner whose interaction is measured |
| Regeneration Solution | Chemical solution (e.g., low pH or high salt buffer) | Removes bound analyte without damaging immobilized ligand |
Instrumentation and Methodologies
SPR Configurations and Imaging Modalities
While the traditional prism-coupled SPR system remains widely used, several advanced configurations have been developed to enhance capabilities and address limitations:
Prism-Coupled SPR Imaging (SPRi): This configuration enables simultaneous monitoring of molecular interactions at multiple spots on the sensor surface, a capability known as multiplexing. By patterning different ligands in a microarray format and using a camera to detect resonance changes across the entire chip surface, researchers can monitor hundreds of interactions in parallel. However, this approach typically provides spatial resolution larger than 10 μm due to optical constraints [3] [1].
SPR Microscopy (SPRM): Using a high numerical aperture oil-immersion objective based on the Kretschmann configuration, SPRM achieves significantly improved spatial resolution of approximately 300 nanometers perpendicular to the propagation direction of surface plasmon waves. This enhanced resolution enables imaging of single nanoparticles, virions, and subcellular structures. A limitation, however, is the formation of parabolic tails in the images along the plasmon propagation direction, which reduces spatial resolution in that dimension [3].
Surface Plasmonic Scattering Microscopy (SPSM): This novel approach directly collects surface plasmon waves scattered by analytes, eliminating interference from propagating surface plasmon waves with long decaying lengths. SPSM achieves diffraction-limited spatial resolutions in all lateral directions in real time and provides high-contrast images capable of resolving single proteins without labels [3].
The following diagram illustrates the core working principle of an SPR instrument based on the Kretschmann configuration:
Kinetic and Affinity Analysis
SPR provides unparalleled capability for quantifying molecular interactions in real time, generating data-rich sensorgrams that track the binding response throughout the experiment:
- Association Phase: When analyte is injected and binds to the immobilized ligand, the response increases as complex formation occurs.
- Equilibrium Plateau: The response stabilizes when the rates of association and dissociation become equal.
- Dissociation Phase: When analyte injection stops and buffer flow resumes, the response decreases as complexes dissociate [1].
These sensorgrams are quantitatively analyzed to determine kinetic parameters:
- Association rate constant (kₐ or k_ass), measured in M⁻¹s⁻¹
- Dissociation rate constant (kd or kdiss), measured in s⁻¹
From these kinetic parameters, the equilibrium dissociation constant (KD), representing the affinity of the interaction, is calculated as KD = k_d/kₐ [4] [2] [1]. The following workflow outlines the key stages of an SPR experiment and the resulting data analysis:
Table 2: Key Parameters Obtained from SPR Analysis
| Parameter | Symbol | Units | Interpretation |
|---|---|---|---|
| Association Rate Constant | kₐ (k_ass) | M⁻¹s⁻¹ | Rate of complex formation; higher values indicate faster binding |
| Dissociation Rate Constant | kd (kdiss) | s⁻¹ | Rate of complex breakdown; higher values indicate faster dissociation |
| Equilibrium Dissociation Constant | K_D | M | Measure of affinity; lower values indicate tighter binding |
| Association Rate Constant | K_A | M⁻¹ | Alternative affinity measure; KA = 1/KD |
Advanced Applications and Current Research Directions
Innovative Applications in Drug Discovery and Biotechnology
SPR technology has become particularly valuable in GPCR drug discovery, where it helps overcome the challenges associated with studying these complex membrane proteins. GPCRs (G protein-coupled receptors) represent one of the most important classes of drug targets but are notoriously difficult to work with due to their instability outside the membrane environment. Recent SPR advances have enabled GPCR analysis through various immobilization strategies, including:
- Native membrane immobilization using whole cells or membrane fragments
- Membrane mimetics such as lipoparticles, liposomes, and nanodiscs
- Stabilized isolated receptors through detergent optimization or protein engineering [5]
In the broader field of biotechnology, SPR has evolved beyond basic binding analysis to enable:
- Single-molecule detection through advanced SPR microscopy techniques
- High-throughput screening of compound libraries in drug discovery
- Biomarker identification and validation for diagnostic applications
- Environmental monitoring of pollutants with sensitivity comparable to traditional chromatography methods but with significantly faster analysis times [6] [3]
Emerging Sensing Materials and Nanostructures
The performance of SPR sensors is being dramatically enhanced through innovations in nanomaterials and sensing interfaces. Research focuses on developing materials that maximize sensitivity, specificity, and stability:
Localized Surface Plasmon Resonance (LSPR) utilizes metal nanoparticles (typically gold or silver) whose collective electron oscillations generate enhanced electromagnetic fields at the nanoscale. LSPR sensors detect analytes through changes in absorption wavelength and can exhibit visible color changes upon target binding, enabling applications in portable and point-of-care diagnostics [6].
Two-dimensional materials such as graphene and transition metal dichalcogenides are being incorporated into SPR sensors to enhance sensitivity through their unique electronic properties and high surface-to-volume ratio.
Bimetallic nanoparticles and metal-organic frameworks (MOFs) are engineered to fine-tune plasmonic properties and introduce additional functionality through their porous structures and selective adsorption characteristics [6].
These material advances, combined with ongoing miniaturization efforts including fiber-based SPR sensors that eliminate the need for prisms, are making SPR technology increasingly accessible for field deployment and point-of-care applications.
Essential Research Reagents and Experimental Considerations
Successful SPR experimentation requires careful selection of reagents and optimization of experimental conditions. The following table outlines key solutions and materials essential for SPR research:
Table 3: Essential Research Reagent Solutions for SPR Experiments
| Reagent/Material | Composition/Type | Function in SPR Experiments |
|---|---|---|
| Sensor Chips | Series S CM5, NTA, SA, etc. (Cytiva) | Provides functionalized surface for ligand immobilization |
| Running Buffer | HBS-EP, PBS-P, etc. with 0.05% Tween 20 | Maintains constant conditions; reduces non-specific binding |
| Immobilization Reagents | Amine coupling kits, thiol coupling reagents | Covalently attaches ligand to sensor surface |
| Regeneration Solutions | Glycine pH 1.5-3.0, NaOH, SDS | Removes bound analyte while maintaining ligand activity |
| System Cleaning Solutions | Desorb 1, Desorb 2, Biadisinfectant | Maintains instrument performance and prevents contamination |
Critical experimental considerations include:
- Temperature control between 4-45°C to maintain biological activity and ensure reproducible kinetic measurements
- Reference surface utilization to subtract systemic artifacts and buffer effects
- DMSO concentration matching when working with small molecule compounds to eliminate solvent effects
- Surface regeneration optimization to ensure complete analyte removal without damaging the immobilized ligand [4]
As SPR technology continues to evolve, its applications are expanding into increasingly complex biological systems and challenging environments. The ongoing development of high-resolution SPR imaging, miniaturized portable systems, and advanced nanomaterials ensures that SPR will remain at the forefront of label-free detection technology, providing critical insights into molecular interactions that drive advances in basic research, drug discovery, and diagnostic applications.
Surface Plasmon Resonance (SPR) has established itself as a cornerstone technology for real-time, label-free analysis of molecular interactions, with profound implications for drug discovery, biosensing, and diagnostic development. The excitation of surface plasmons, however, requires a precise optical configuration to overcome the momentum mismatch between incident light and surface plasmon waves. Among the various coupling strategies, the Kretschmann configuration has emerged as the predominant and most widely adopted experimental setup in commercial and research instruments. This whitepaper provides an in-depth technical examination of the Kretschmann configuration, detailing its fundamental operating principles, theoretical underpinnings, and practical implementation. By synthesizing current research and quantitative performance data, this guide serves as a comprehensive resource for researchers and scientists seeking to leverage this powerful technology for advanced molecular interaction analysis.
Surface Plasmon Resonance (SPR) is an optical phenomenon occurring at the interface between a metal and a dielectric, where incident light couples with collective oscillations of free electrons (plasmons) in the metal. This coupling results in a characteristic dip in the reflected light intensity at a specific resonance condition, which is exquisitely sensitive to changes in the refractive index within the evanescent field—typically extending 100-200 nm from the metal surface [7]. This physical principle forms the basis for a powerful analytical technique that enables real-time, label-free monitoring of biomolecular interactions, including antibody-antigen binding, protein-protein interactions, and DNA hybridization [4].
The Kretschmann configuration, first described in 1968, provides the most practical and efficient method for exciting surface plasmons in the laboratory setting [8]. In this arrangement, a thin metal film (typically gold or silver) is directly deposited onto the base of a high-refractive-index prism. When polarized light undergoes total internal reflection (TIR) within the prism, it generates an evanescent wave that penetrates through the metal film, exciting surface plasmons at the outer metal-dielectric interface under precise resonance conditions [7]. This configuration has largely superseded the earlier Otto configuration, where an air gap exists between the prism and the metal layer, due to its superior efficiency and experimental convenience, particularly for applications involving liquid samples [8].
Theoretical Foundations
Fundamental Physics and Resonance Condition
The excitation of surface plasmons in the Kretschmann configuration relies on the principle of attenuated total reflection (ATR). The core physical requirement is the matching of momentum between the incident photon and the surface plasmon. This is achieved by utilizing the evanescent wave generated during total internal reflection to provide the necessary momentum boost.
The resonance condition is mathematically described by equating the wave vector of the incident light to that of the surface plasmon polariton:
kSP = kevan,∥
where kSP represents the surface plasmon wave vector and kevan,∥ represents the parallel component of the evanescent wave vector [7].
Expanding this fundamental equation provides the practical resonance condition:
$$\frac{2\pi}{\lambda} np \sin(\theta{SPR}) = \frac{2\pi}{\lambda} \sqrt{\frac{\epsilonm \epsilond}{\epsilonm + \epsilond}}$$
In this equation:
- λ is the wavelength of the incident light
- np is the refractive index of the prism
- θSPR is the resonance angle of incidence
- εm is the dielectric constant of the metal
- εd is the dielectric constant of the dielectric medium (analyte solution) [9] [7]
This relationship demonstrates that any alteration in the dielectric constant of the adjacent medium (εd), such as through molecular adsorption or binding events, directly affects the resonance condition, enabling quantitative detection of molecular interactions.
Visualizing the Core Concept
The following diagram illustrates the fundamental components and light path in the Kretschmann configuration:
Figure 1: Core Components of the Kretschmann Configuration. P-polarized light enters the prism and reflects off the metal-coated base at an angle θi. At the resonance angle θSPR, the evanescent wave couples with electron oscillations in the metal film, generating surface plasmons and causing a characteristic dip in reflected light intensity.
Implementation and Instrumentation
Core Components and Experimental Setup
A functional Kretschmann-based SPR instrument requires several integrated optical and fluidic components:
- Light Source: A polarized laser diode (typically λ = 650 nm) provides coherent, monochromatic light essential for precise resonance measurements [7].
- High-Index Prism: Commonly made of BK7 glass, serving as the TIR medium and optical coupler.
- Metallic Film: A thin film (~50 nm gold) vacuum-sputtered or thermally evaporated onto the prism base [7].
- Detection System: A position-sensitive detector (e.g., photodiode array or CCD camera) measures the intensity of reflected light as a function of angle [8] [7].
- Microfluidic System: Provides controlled delivery of analyte solutions to the sensor surface, enabling real-time binding measurements [4].
In angular interrogation systems, the incident angle (θ) is varied while monitoring reflected intensity at a fixed wavelength. The resulting plot of reflectance versus incident angle displays a sharp minimum at θSPR, which shifts in response to changes in the refractive index at the metal surface due to molecular binding events [7].
Advanced Configurations and Material Enhancements
While the basic Kretschmann configuration uses a single metal layer, research has demonstrated that enhanced performance can be achieved through sophisticated multilayer structures. These advanced configurations often incorporate two-dimensional (2D) materials to improve sensitivity and provide functional groups for biomolecule immobilization.
Table 1: Performance Comparison of Kretschmann Configuration with Different Material Stacks for DNA Sensing
| Configuration Structure | Sensitivity (deg/RIU) | Detection Accuracy | Quality Factor (RIU⁻¹) | Key Features |
|---|---|---|---|---|
| Conventional Ag | Baseline | Baseline | Baseline | Reference structure |
| Ag-Graphene | Moderate improvement | Moderate improvement | Moderate improvement | Graphene enhances biomolecule adhesion via π-π stacking |
| Ag-Graphene-Au-WS₂-MoS₂ | 203 | 1.13 | 28.31 | Optimal performance with bimetallic layers and 2D materials [10] |
Recent studies have explored structures featuring a sandwiched graphene layer between silver and gold, topped with transition metal dichalcogenides (TMDCs) like WS₂ and MoS₂. This arrangement leverages the high sensitivity of silver while protecting it from oxidation with a thin gold layer, and utilizes 2D materials to significantly enhance sensitivity through their high surface-to-volume ratio and strong light-matter interaction [10].
Experimental Protocols and Applications
Generalized Experimental Workflow
The following diagram outlines the standard workflow for conducting an SPR experiment using the Kretschmann configuration:
Figure 2: Standard SPR Experimental Workflow. The process begins with surface preparation and ligand immobilization, followed by real-time monitoring of analyte binding and dissociation, concluding with data analysis to extract kinetic parameters.
Detailed Methodology: DNA Detection Sensor Fabrication
The following protocol, adapted from recent research, details the fabrication of a high-sensitivity SPR biosensor for single-stranded DNA (ssDNA) detection using 2D materials [10]:
- Substrate Preparation: Begin with a BK-7 glass prism thoroughly cleaned with piranha solution and dried under nitrogen gas.
- Metal Deposition: Deposit a 44 nm silver layer onto the prism base using vacuum thermal coating procedures.
- Graphene Transfer: Transfer a monolayer of graphene synthesized via chemical vapor deposition (CVD) onto the silver layer.
- Gold Coating: Deposit a 4 nm gold layer using thermal coating techniques over the graphene monolayer.
- TMDC Application: Transfer bilayer WS₂ and monolayer MoS₂ (grown by CVD method) sequentially onto the gold layer.
- Functionalization: Immobilize monolayer ssDNA probes onto the sensing surface for specific target capture.
- Measurement: Introduce analyte solutions containing complementary DNA strands while monitoring resonance shifts.
This configuration has demonstrated a sensitivity of 203 deg/RIU with a detection accuracy of 1.13 and quality factor of 28.31 RIU⁻¹, significantly outperforming conventional single-metal sensors [10].
The Scientist's Toolkit: Essential Research Reagents
Table 2: Key Research Reagents and Materials for Kretschmann Configuration Experiments
| Component | Specifications | Function/Purpose |
|---|---|---|
| Sensor Chips | Cytiva Series S chips (e.g., CM5) | Commercial substrates with specialized coatings for biomolecule immobilization [4] |
| Prism Material | BK-7 glass (n = 1.515) or SF11 glass (n = 1.778) | High-index optical element for total internal reflection [10] [9] |
| Plasmonic Metals | Gold (50 nm) or Silver (47 nm) | Surface plasmon generation; gold offers stability, silver provides higher sensitivity [9] [7] |
| 2D Enhancement Materials | Graphene, MoS₂, WS₂ | Sensitivity enhancers and biomolecule adhesion layers [10] |
| Running Buffer | HBS-EP (0.01 M HEPES, 0.15 M NaCl, 0.005% v/v Tween 20) | Maintains constant pH and ionic strength; detergent minimizes non-specific binding [4] |
| Immobilization Chemistry | Carbodiimide (EDC/NHS) coupling | Covalent attachment of ligands to sensor surface [4] |
| Regeneration Solutions | Glycine-HCl (pH 2.5-3.0), NaOH (10-100 mM) | Removes bound analyte without damaging immobilized ligand [4] |
Performance Analysis and Optimization Strategies
Critical Performance Parameters
The performance of Kretschmann-configuration SPR sensors is quantified through several key parameters:
- Sensitivity (S): Defined as the shift in resonance signal per unit change in refractive index, typically measured in deg/RIU (angular interrogation) or nm/RIU (wavelength interrogation). Higher sensitivity enables detection of smaller molecular interactions or lower analyte concentrations [10].
- Detection Accuracy (D.A.): Related to the sharpness of the resonance dip, affecting the precision with which the resonance minimum can be determined [10].
- Quality Factor (Q.F.): A comprehensive metric considering both sensitivity and resonance width, providing a balanced assessment of overall sensor performance [10].
- Full Width at Half Maximum (FWHM): The angular or spectral width of the resonance curve at half its minimum reflectance, with narrower dips enabling more precise resonance tracking [11].
- Figure of Merit (FoM): Often defined as Sensitivity/FWHM, providing a normalized performance measure for comparing different sensor configurations [11].
Comparative Performance Across Applications
Table 3: Performance Metrics of Kretschmann Configuration in Various Sensing Applications
| Application | Sensor Structure | Sensitivity | Detection Limit | Key Findings |
|---|---|---|---|---|
| ssDNA Detection | Ag/Graphene/Au/WS₂/MoS₂ | 203 deg/RIU | N/A | Bimetallic layer with 2D materials maximizes sensitivity [10] |
| Cyanide Ion Sensing in Water | Ag/MAFBP-silica matrix | 6.9 nm/μM | 0.14 μM | Porphyrin-based recognition provides high selectivity below WHO limits [9] |
| Voltage Sensing | Bimetallic Ag/Au | Variable with mechanism | N/A | FoM enhancement of 1.34-25x over single metal layers [11] |
Optimization Strategies
Maximizing SPR sensor performance requires careful optimization of multiple parameters:
- Metal Selection and Thickness: Silver provides higher sensitivity but oxidizes easily; gold offers better chemical stability but lower sensitivity. Optimal metal thickness ranges between 45-55 nm [9] [7].
- Wavelength Considerations: Longer wavelengths generally provide higher sensitivity but broader resonance dips, requiring a balance based on application requirements.
- 2D Material Integration: Graphene enhances biomolecule adhesion through π-π stacking, while TMDCs (MoS₂, WS₂) enhance sensitivity through their high surface-to-volume ratio and exceptional optical properties [10].
- Temperature Control: Maintaining stable temperature (±0.03°C) is critical as refractive index is highly temperature-dependent [4].
Current Research and Future Perspectives
The Kretschmann configuration continues to evolve through ongoing research efforts. Recent investigations have explored hybrid configurations that combine Kretschmann arrangements with other photonic phenomena, such as the experimental demonstration of simultaneous lossy mode resonances (LMRs) and SPR generation using indium tin oxide (ITO) coatings [12]. These hybrid approaches offer new opportunities for multimodal sensing and enhanced performance.
In biosensing applications, researchers are developing increasingly sophisticated functionalization strategies to improve specificity and reduce non-specific binding. The integration of synthetic recognition elements such as malononitrile-appended free-base di-fused porphyrin (MAFBP) for cyanide ion detection demonstrates how molecular engineering can create highly selective sensors for environmental monitoring [9].
Future directions in Kretschmann-configuration SPR research include the development of high-throughput SPR imaging (SPRi) systems for parallel analysis of hundreds of interactions [8], miniaturization of system components for point-of-care applications, and integration with complementary analytical techniques such as electrochemistry [7] and mass spectrometry. These advances will further solidify the central role of the Kretschmann configuration in interfacial analysis and molecular interaction studies.
The Kretschmann configuration remains the gold standard for SPR excitation due to its robust design, experimental versatility, and exceptional sensitivity to interfacial phenomena. While its fundamental principles have remained consistent since its inception, ongoing innovations in nanomaterials, detection methodologies, and surface chemistry continue to expand its capabilities and applications. For researchers in drug development and biomedical sciences, mastery of this configuration provides a powerful tool for unraveling complex molecular interactions, characterizing binding kinetics, and developing novel diagnostic assays. As SPR technology continues to evolve, the Kretschmann configuration will undoubtedly maintain its position as the foundational architecture for surface plasmon resonance research and applications.
Surface Plasmon Resonance (SPR) biosensors represent a cornerstone of modern analytical science, enabling the real-time, label-free investigation of biomolecular interactions. The operational principle of these sensors is rooted in the sophisticated interplay between evanescent waves and collective electron oscillations, known as plasmons, at metal-dielectric interfaces [13] [14]. This physical phenomenon provides an exceptionally sensitive probe for detecting changes in the local refractive index, which forms the basis for quantifying biomolecular binding events [15].
The significance of SPR technology extends across multiple disciplines, including molecular biology, pharmaceutical research, and medical diagnostics [16]. Its ability to provide real-time binding data and kinetic characterization has made it indispensable in drug discovery workflows and clinical applications, facilitating the detection of targets ranging from proteins and nucleic acids to viruses and whole cells [15] [14]. This technical guide examines the fundamental principles of evanescent waves and plasmon oscillations, their roles in SPR signal transduction, and their practical applications within a broader research context.
Fundamental Concepts: Evanescent Waves and Plasmon Oscillations
The Origin and Nature of Evanescent Waves
Evanescent waves form the foundational optical component of SPR biosensing. They arise under conditions of total internal reflection (TIR), which occurs when light traveling through an optically dense medium (such as a glass prism) strikes an interface with a less dense medium (such as a liquid sample) at an angle greater than the critical angle [14]. While the majority of the light energy is reflected back into the denser medium, a portion of the wave's energy penetrates the interface as an evanescent field.
This evanescent wave is characterized by its exponential decay with increasing distance from the interface, typically extending only a few hundred nanometers into the less dense medium [13]. It does not propagate energy across the interface but rather stores energy in a standing wave pattern parallel to the interface. The intensity (I) of this field at a distance z from the interface is described by:
I(z) = I₀e^(-z/d_p)
where I₀ is the intensity at the interface and d_p is the penetration depth, typically ranging from 100-300 nm for visible light, which is commensurate with the size of many biomolecules [13].
Plasmon Oscillations: Collective Electron Behavior
Surface plasmons are coherent oscillations of free electrons at the boundary between a metal (typically gold or silver) and a dielectric medium [6] [14]. These collective electron excitations are classified into two primary types based on their confinement:
- Propagating Surface Plasmons: Also known as surface plasmon polaritons (SPPs), these electromagnetic waves propagate along the metal-dielectric interface over distances of micrometers before decaying [14].
- Localized Surface Plasmons (LSP): These non-propagating plasmons are confined to metallic nanostructures or nanoparticles with dimensions smaller than the wavelength of incident light [6]. LSPR sensors detect changes through shifts in absorption wavelength caused by analyte interactions with nanoparticle surfaces.
The resonance condition for exciting surface plasmons is highly sensitive to the dielectric constant of both the metal and the adjacent medium, making it an exquisite probe for detecting molecular binding events at the metal surface [14].
Signal Generation: The Coupling Mechanism
The Resonance Condition
The core mechanism of SPR biosensing relies on coupling light energy to surface plasmons via the evanescent field [14]. This coupling occurs under precise resonance conditions where the wavevector of the incident light matches that of the surface plasmon. For p-polarized light incident on a metal surface, the wavevector component parallel to the interface (kₓ) is given by:
kₓ = (ω/c)√ε_g × sinθ
where ω is the angular frequency of light, c is the speed of light, ε_g is the dielectric constant of the prism material, and θ is the angle of incidence [14].
The surface plasmon wavevector (k_sp) is described by:
ksp = (ω/c)√(εm εd)/(εm + ε_d)
where εm and εd are the dielectric constants of the metal and dielectric medium, respectively [14]. Resonance occurs when kₓ = k_sp, resulting in a transfer of energy from the incident photons to surface plasmons, observed as a sharp dip in the intensity of reflected light at a specific angle of incidence [14].
Transduction of Molecular Binding Events
When biomolecules bind to the functionalized metal surface, they alter the local refractive index within the evanescent field penetration depth [13] [14]. This change modifies the resonance condition, shifting the angle, wavelength, or intensity at which resonance occurs [15]. The magnitude of this shift is directly proportional to the mass concentration of bound analyte, enabling quantitative measurements of binding interactions in real time without requiring fluorescent or radioactive labels [13].
Table 1: Key Parameters in SPR Signal Generation
| Parameter | Symbol | Typical Values/Range | Impact on Sensing |
|---|---|---|---|
| Penetration Depth | d_p | 100-300 nm | Determines sensing volume and size compatibility with target analytes |
| Resonance Angle | θ_RES | Varies with setup | Shifts upon molecular binding (0.01° can be significant) |
| Metal Film Thickness | t_m | ~50 nm (gold) | Affects resonance sharpness and coupling efficiency |
| Refractive Index Change | Δn | 10⁻⁶ to 10⁻³ RIU | Directly correlates with bound analyte mass |
Quantitative Performance Metrics of SPR Biosensors
The analytical performance of SPR biosensors is characterized by several key metrics that determine their suitability for specific applications. Understanding these parameters is essential for method development and data interpretation in research settings.
Table 2: Performance Metrics of SPR Biosensing Platforms
| Performance Metric | Typical Range for SPR | LSPR Sensors | Impact on Applications |
|---|---|---|---|
| Sensitivity | Varies by configuration [6] | High nanoscale sensitivity [6] | Determines lowest detectable analyte concentration |
| Detection Limit | ~1 pg/mm² [15] | ppb level for trace substances [6] | Crucial for low-abundance biomarker detection |
| Real-time Monitoring | Yes (millisecond resolution) [16] | Yes [6] | Enables kinetic profiling of molecular interactions |
| Label-free Operation | Yes [13] [16] | Yes [6] | Preserves native biomolecule structure and function |
| Multiplexing Capability | Yes (with imaging systems) [13] | Yes (through nanoparticle encoding) [6] | Increases throughput for screening applications |
Experimental Methodology: SPR Biosensor Setup
Standard Kretschmann Configuration
The most widely implemented experimental setup for SPR biosensing is the Kretschmann configuration, which uses a prism coupler to facilitate the evanescent wave excitation of surface plasmons on a thin metal film [14]. The following protocol details the essential steps for establishing a functional SPR biosensor:
Materials and Reagents:
- High-index glass prism (e.g., SF10 glass, ε_g)
- ~50 nm gold film deposition on prism surface
- Polarizer to generate p-polarized light
- Flow cell system for sample delivery
- Optical detection system (CCD camera or photodiode array)
- Phosphate-buffered saline (PBS), pH 7.4 for running buffer
- Functionalization reagents: alkanethiols for self-assembled monolayers (SAMs)
- Capture molecules: antibodies, aptamers, or receptors specific to target analyte
Instrument Setup:
- Mount the metal-coated prism on a rotational stage or goniometer capable of precise angular control (±0.001°).
- Align the optical path such that a collimated, p-polarized light beam strikes the metal-prism interface at a variable angle.
- Position the detector to capture the intensity profile of reflected light across a range of incident angles.
- Connect the flow cell to the metal surface, ensuring a leak-free seal.
- Integrate fluid handling system with precision pumps for buffer and sample delivery.
Surface Functionalization Protocol:
- Clean the gold surface with oxygen plasma treatment or piranha solution (Caution: highly corrosive).
- Form a self-assembled monolayer (SAM) by incubating with 1-10 mM alkanethiol solution in ethanol for 12-24 hours.
- Rinse thoroughly with ethanol and running buffer to remove unbound thiols.
- Activate the SAM surface using EDC/NHS or similar chemistry for covalent attachment of capture molecules.
- Immobilize capture molecules (antibodies, aptamers, etc.) at optimal density (typically 1-10 ng/mm²).
- Block remaining active sites with inert proteins (BSA, casein) or ethanolamine to minimize nonspecific binding.
Measurement Procedure:
- Establish a stable baseline with running buffer flowing at constant rate (typically 10-50 μL/min).
- Acquire reflectivity scans across the angular range to determine the initial resonance angle.
- Introduce sample solution containing analyte for a specified association phase.
- Monitor the angular shift in real-time as analytes bind to the surface.
- Replace with running buffer to monitor dissociation of bound complexes.
- Regenerate the surface if needed using mild acidic or basic conditions to remove bound analytes without damaging the capture layer.
Alternative Configurations and Recent Advances
While the Kretschmann configuration remains predominant, several alternative setups have been developed to address specific application needs:
- Grating-Coupled SPR: Uses periodic nanostructures instead of a prism to couple incident light to surface plasmons, enabling more compact designs [14].
- Optical Fiber SPR: Integrates SPR sensing with optical fibers, facilitating miniaturization and remote sensing capabilities [13] [6].
- Imaging SPR (SPRi): Enables spatially resolved monitoring of multiple interactions simultaneously through CCD-based detection [13].
- Localized SPR (LSPR): Utilizes nanostructured metal surfaces or nanoparticles rather than planar films, offering enhanced sensitivity for certain applications and simplified optical setups [6].
Research Reagent Solutions for SPR Biosensing
Successful implementation of SPR biosensing requires carefully selected reagents and materials optimized for the specific experimental goals. The following table details essential components and their functions in typical SPR workflows.
Table 3: Essential Research Reagents for SPR Biosensor Development
| Reagent/Material | Function/Purpose | Examples/Specifications |
|---|---|---|
| Gold Film (~50 nm) | Plasmon-active metal surface | Evaporation/sputtered on prism or glass substrate |
| Alkanethiols | Self-assembled monolayer formation | 11-mercaptoundecanoic acid (11-MUA) for carboxyl groups |
| Coupling Agents | Covalent immobilization of ligands | EDC/NHS chemistry for amine coupling |
| Capture Molecules | Target-specific recognition | Antibodies, aptamers, receptors [14] |
| Aptamers | Synthetic oligonucleotide recognition elements | 20-200 nucleotides; high affinity/specificity to targets [14] |
| Blocking Agents | Reduction of nonspecific binding | BSA, casein, ethanolamine, surfactants |
| Regeneration Solutions | Surface reset without damage | Glycine-HCl (pH 2.0-3.0), NaOH (10-100 mM) |
| 2D Nanomaterials | Sensitivity enhancement | Graphene, transition metal dichalcogenides [6] [14] |
| Microfluidic Components | Precise fluid delivery | Flow cells, tubing, precision syringe pumps |
Signaling Pathways and Experimental Workflows
The following diagrams visualize key signaling pathways and experimental workflows in SPR biosensing, created using Graphviz DOT language with high color contrast for clarity.
SPR Signal Transduction Pathway
SPR Experimental Workflow
Applications in Drug Discovery and Medical Diagnostics
The unique capabilities of SPR biosensors have established them as invaluable tools in pharmaceutical research and clinical diagnostics, with several well-defined application areas:
Drug Discovery and Development
SPR biosensors have revolutionized multiple stages of the drug discovery pipeline through their ability to provide detailed kinetic information on molecular interactions [16]. Key applications include:
- Hit Identification and Validation: SPR enables high-throughput screening of compound libraries against therapeutic targets, rapidly identifying promising lead compounds with the desired binding characteristics [16].
- Affinity and Kinetics Optimization: By providing real-time data on association (kon) and dissociation (koff) rates, SPR guides medicinal chemistry efforts to optimize drug-target interactions [14].
- Fragment-Based Drug Design (FBDD): SPR's sensitivity allows detection of weak interactions between low molecular weight fragments and target proteins, providing starting points for drug development [16].
- Antibody Characterization: SPR is extensively used to characterize monoclonal antibodies, determining affinity, specificity, and cross-reactivity profiles critical for therapeutic antibody development [14].
Medical Diagnostics and Biomarker Detection
SPR biosensors have demonstrated significant potential in clinical diagnostics through their ability to detect disease-specific biomarkers in complex biological matrices [13] [15]. Notable applications include:
- Infectious Disease Detection: SPR platforms have been developed to detect viral particles (including SARS-CoV-2), bacterial pathogens, and associated antibodies with clinical-level sensitivity and specificity [14].
- Cancer Biomarker Monitoring: SPR biosensors can identify circulating tumor cells, exosomes, and protein biomarkers at clinically relevant concentrations, enabling early cancer detection and monitoring [15].
- Cardiovascular Disease Markers: Cardiac-specific biomarkers such as troponins and CRP can be detected at diagnostically significant levels using optimized SPR assays [15].
The translational potential of SPR in medical applications continues to expand with advancements in nanomaterial-enhanced sensitivity, chip-scale multiplexing, and portable point-of-care designs that bridge the gap between fundamental research and clinical implementation [13].
The sophisticated interplay between evanescent waves and plasmon oscillations forms the physical foundation of SPR biosensing technology. This coupling mechanism enables highly sensitive, label-free detection of molecular interactions in real time, making SPR an indispensable tool in both basic research and applied pharmaceutical development. As material science and optical engineering continue to advance, SPR platforms are evolving toward greater sensitivity, miniaturization, and integration with complementary technologies like microfluidics and artificial intelligence [13]. These developments promise to further expand the applications of SPR in drug discovery, medical diagnostics, and beyond, solidifying its role as a cornerstone analytical technique in the molecular sciences.
Surface Plasmon Resonance (SPR) is an optical technique that enables the real-time, label-free analysis of biomolecular interactions [4] [17]. At the heart of any SPR experiment lies the sensorgram, a dynamic plot that provides a visual representation of the interaction lifecycle between a ligand immobilized on a sensor chip and an analyte in solution [18] [19]. By monitoring this interaction in real time, researchers can extract crucial parameters, including the kinetics, affinity, and specificity of the binding event [18].
The fundamental principle of SPR involves the excitation of surface plasmons in a thin metal film (typically gold) under conditions of total internal reflection [4]. The SPR signal is exquisitely sensitive to changes in the refractive index at the sensor surface. When an analyte binds to the immobilized ligand, the resulting increase in mass at the surface causes a proportional change in the refractive index, which is detected as a shift in the resonance angle or wavelength [18] [4]. This shift is plotted against time to generate the sensorgram, with the response measured in Resonance Units (RU) [4]. One RU corresponds to approximately 1 pg of protein per mm², making SPR a highly sensitive technique for monitoring binding events [4].
The Anatomy of a Sensorgram
A typical sensorgram is composed of five distinct phases, each providing specific information about the molecular interaction and the state of the sensor system [18]. The following diagram illustrates the complete process of how an SPR instrument converts a binding event into a sensorgram.
Figure 1: From Binding Event to Sensorgram: This diagram illustrates the optical principle of SPR detection. A binding event on the gold film changes the refractive index, causing a shift in the SPR dip (resonance angle). The instrument detects this shift and converts it in real-time into the sensorgram plot.
The Five Key Phases
Baseline: This initial phase establishes the system's stability before analyte injection [18] [19]. A stable, flat baseline, achieved by flowing a running buffer (e.g., phosphate-buffered saline or HEPES-NaCl) over the sensor surface, is critical for accurate measurements [18]. Significant baseline drift can indicate system contamination, buffer mismatch, or temperature fluctuations, necessitating cleaning of the fluidics or sensor chip [19].
Association: This phase begins with the injection of the analyte over the ligand-functionalized surface [18] [19]. The binding of analyte to ligand causes an increase in mass at the surface, leading to a positive shift in the SPR response and an upward curve on the sensorgram [4]. The shape of the association curve is governed by the association rate constant (kon), the concentration of the analyte, and the density of the available ligand [20].
Steady-State (Equilibrium): This is not always achieved but is represented by a plateau in the sensorgram where the net rate of binding becomes zero; the number of analyte molecules binding to the ligand equals the number dissociating [18]. The response level at this plateau can be used to determine the equilibrium dissociation constant (KD) [20].
Dissociation: Initiated by switching back to a continuous flow of buffer, this phase monitors the unbinding of the analyte from the ligand, resulting in a decrease in the SPR response [18] [19]. The slope of this downward curve is determined by the dissociation rate constant (koff) and reflects the stability of the complex; a slower dissociation indicates a more stable complex [18] [20].
Regeneration: To prepare the sensor surface for a new experiment, a regeneration buffer (often a low-pH solution like glycine) is injected to disrupt the ligand-analyte interaction without permanently damaging the immobilized ligand [18] [19]. A successful regeneration resets the SPR response to the original baseline, confirming the surface is ready for reuse [18]. Finding regeneration conditions that are effective yet gentle is often an empirical process [21].
Table 1: Key Parameters Derived from Sensorgram Analysis
| Parameter | Symbol | Definition | Significance in Drug Discovery |
|---|---|---|---|
| Association Rate Constant | kon (M-1s-1) | Speed at which analyte binds to ligand | A fast on-rate can lead to a quicker onset of pharmacological effect [20] |
| Dissociation Rate Constant | koff (s-1) | Speed at which the complex dissociates | A slow off-rate (long residence time) can confer durability and prolonged efficacy [20] |
| Equilibrium Dissociation Constant | KD (M) = koff / kon | Affinity; concentration of analyte at which half the ligand is bound | Lower KD indicates higher affinity, potentially allowing for lower dosing [20] |
| Maximum Response | Rmax (RU) | Theoretical response at saturating analyte concentration | Used to validate the binding model and calculate binding stoichiometry [21] |
Quantitative Analysis of Sensorgram Data
Kinetic and Affinity Analysis
The primary goal of sensorgram analysis is to extract the kinetic rate constants (kon and koff) and the equilibrium affinity (KD). This is achieved by fitting the sensorgram data to a suitable binding model [18]. The most fundamental model is the 1:1 Langmuir binding model, which describes a simple bimolecular interaction [20].
The analysis requires data from a series of analyte injections at different concentrations. The set of sensorgrams, comprising association and dissociation phases for all concentrations, is globally fitted to the binding model. This process simultaneously determines the kon and koff values that best describe the entire dataset [18]. The KD is then calculated as the ratio koff/kon [20]. For interactions that rapidly reach equilibrium, the KD can also be determined from a steady-state analysis by plotting the equilibrium response versus analyte concentration and fitting to a binding isotherm [21] [20].
Experimental Design and Methodologies
A. Immobilization Strategies
The first critical step in any SPR experiment is the stable immobilization of the ligand to the sensor chip while maintaining its biological activity. The choice of immobilization chemistry depends on the nature of the ligand and the interaction being studied.
- Covalent Coupling: A standard method involves using a CM5 dextran chip and amine-coupling chemistry (NHS/EDC) to form stable amide bonds with primary amines on the ligand [21]. While robust, this can lead to heterogeneous attachment orientations.
- Capture Methods: These techniques offer oriented immobilization, which can enhance binding activity and consistency. Common methods include:
The immobilization level must be optimized. Too high a density can cause mass transport limitation, where the rate of analyte diffusion to the surface becomes slower than the binding rate itself, distorting the kinetic data [19]. A maximum response (Rmax) of around 100 RU is often preferred for kinetic measurements [21].
B. Experimental Protocol for Kinetic Characterization
The following workflow outlines a standard experiment to determine the kinetic parameters of an antibody-antigen interaction, using a Protein A chip for antibody capture.
- Surface Preparation: Dock the sensor chip and prime the instrument with running buffer (e.g., HEPES Buffered Saline with 0.05% Tween 20, pH 7.4) [4] [21].
- Ligand Capture: Inject a diluted antibody solution over a single flow cell on the Protein A chip to achieve a capture level of ~50-100 RU.
- Analyte Injection: Inject a series of antigen concentrations (e.g., a 3-fold dilution series spanning a range above and below the expected KD) over the captured antibody surface and a reference surface. Each injection should have a sufficient association time (e.g., 3-5 minutes) to monitor binding.
- Dissociation Monitoring: Switch to buffer flow and monitor the dissociation of the antigen for a sufficient time (e.g., 5-10 minutes) to reliably determine the koff.
- Surface Regeneration: Inject a brief pulse of regeneration buffer (e.g., 10 mM Glycine, pH 2.0) to remove the antigen and the captured antibody, readying the surface for the next cycle. Repeat steps 2-5 for each antigen concentration [18] [19].
- Data Analysis: Subtract the reference flow cell sensorgram to correct for bulk refractive index changes and non-specific binding. Globally fit the resulting set of sensorgrams to a 1:1 binding model to obtain kon, koff, and KD [18].
Table 2: Essential Research Reagents and Materials for SPR
| Item | Function / Description | Example Products / Components |
|---|---|---|
| Sensor Chips | Solid support with a gold film and functional matrix for ligand immobilization. | CM5 (carboxymethylated dextran), NTA (Ni2+ chelation), SA (streptavidin), Protein A [4] [21] |
| Running Buffer | Continuous flow medium to maintain stable baseline and carry analyte. | PBS, HEPES-NaCl, often with surfactant (e.g., 0.05% Tween 20) [18] [4] |
| Regeneration Buffers | Solutions to remove bound analyte without damaging the immobilized ligand. | Low pH (10-100 mM Glycine-HCl, pH 2.0-3.0), high salt (2 M NaCl), mild acid/base [18] [21] |
| Capture Ligands | For oriented immobilization of the molecule of interest. | Protein A, Protein G, Streptavidin, His-tag antibodies [21] |
| Instrument Cleaners | Solutions to remove contaminants from the fluidics system. | Desorb 1 (SDS solution), Desorb 2 (acidic solution), Biadisinfectant [4] |
Advanced Applications and Troubleshooting
Advanced Applications in Biopharmaceutical Research
SPR has evolved beyond simple 1:1 interaction analysis. The P4SPR and Biacore T200 instruments exemplify systems that integrate advanced fitting software for complex models [18] [4]. High-throughput SPR (HT-SPR), as implemented in platforms like the Carterra LSA, enables the simultaneous kinetic screening of thousands of interactions, dramatically accelerating therapeutic discovery timelines [20].
Advanced applications include:
- Glycosylation Characterization: SPR can be used to analyze the glycosylation profiles of monoclonal antibodies (mAbs) in crude samples by measuring their differential binding to Fcγ receptors, providing critical quality attribute data during bioprocessing [22].
- Small Molecule Screening: Despite the challenge of low mass change, SPR is widely used in fragment-based drug discovery to identify and characterize the binding of low molecular weight (<500 Da) compounds to protein targets [21] [23].
- Medical Diagnostics: SPR biosensors are being developed for the direct detection of clinically relevant analytes like nucleic acids, viruses, bacteria, and exosomes from complex biological matrices [15].
Common Sensorgram Issues and Solutions
Even well-designed experiments can encounter issues. The table below summarizes common problems and their solutions.
Table 3: Troubleshooting Common Sensorgram Anomalies
| Problem | Potential Causes | Recommended Solutions |
|---|---|---|
| Baseline Drift | Contaminated buffer or fluidics; unstable temperature; deteriorating sensor surface [19] | Clean fluidics with recommended cleaners; use fresh, filtered buffer; ensure temperature stability [4] [19] |
| Low Binding Signal | Analyte or ligand concentration too low; insufficient ligand immobilization; low affinity interaction [19] | Increase analyte concentration; optimize ligand immobilization level to increase density [21] [19] |
| Non-Specific Binding (NSB) | Analyte interacting with the chip surface rather than the ligand; impurities in sample [19] | Include a reference surface; use a different chip chemistry; add a surfactant to the buffer; purify the analyte sample [4] [19] |
| Mass Transport Limitation | Ligand density is too high; flow rate is too low [19] | Reduce the level of immobilized ligand; increase the flow rate during analyte injection [21] |
| Irregular Curve Shapes | Inhomogeneous ligand; aggregation; incorrect binding model | Check ligand purity and monodispersity; test different binding models (e.g., heterogeneous ligand) |
The SPR sensorgram is a powerful and information-rich data representation that is fundamental to modern biomolecular interaction analysis. A deep understanding of its phases—baseline, association, steady-state, dissociation, and regeneration—is essential for designing robust experiments and interpreting kinetic and affinity data accurately. Mastery of the sensorgram allows researchers in drug development and basic research to elucidate the mechanisms of molecular interactions, driving the discovery and optimization of novel therapeutic agents. As SPR technology continues to advance, with improvements in throughput, sensitivity, and integration with other analytical methods, the sensorgram will remain a central tool for characterizing the intricate dynamics of biological systems.
Surface Plasmon Resonance (SPR) technology represents a cornerstone technique in modern biochemical analysis, enabling the direct, label-free observation of biomolecular interactions in real time. This capability is critical for applications ranging from fundamental biological research to pharmaceutical development, where understanding the precise kinetics of interactions is paramount. This whitepaper delves into the core advantages of SPR, with a particular focus on its capacity to detect transient interactions often missed by traditional endpoint assays. Supported by contemporary research and experimental data, we illustrate how SPR provides unparalleled insights into binding kinetics, affinities, and specificities, thereby framing its role within the broader fundamentals of SPR research.
Surface Plasmon Resonance (SPR) is a powerful, label-free biosensing technology that enables the real-time monitoring of biomolecular interactions [24]. The fundamental principle involves detecting changes in the refractive index on a sensor chip surface, which occur when an analyte binds to an immobilized ligand. This allows for the direct measurement of binding events without the need for fluorescent or radioactive labels, preserving the native state of the interacting molecules [25]. The real-time output of an SPR biosensor is a sensorgram, a plot of response units (RU) against time, which provides a rich dataset on the progression of association and dissociation phases of an interaction.
Within the context of foundational SPR research, the technology's value is rooted in its ability to provide not just qualitative (yes/no binding) but also quantitative data. It can directly measure the association rate constant (kₐ), dissociation rate constant (kd), and calculate the equilibrium dissociation constant (KD), offering a comprehensive kinetic and thermodynamic profile of the interaction under study [26]. This real-time, kinetic capability distinguishes SPR from traditional endpoint assays and is crucial for a deep understanding of dynamic molecular processes in fields like immunology, proteomics, and drug discovery [27].
Core Advantages: Real-Time and Label-Free Detection
The dual advantages of being both real-time and label-free form the bedrock of SPR's utility in advanced research. These characteristics provide distinct benefits over conventional techniques.
The Critical Importance of Real-Time Kinetics
Real-time monitoring fundamentally changes how researchers perceive and characterize molecular interactions. Traditional endpoint assays provide a single snapshot measurement after incubation and wash steps, making them susceptible to false-negative results for interactions with fast dissociation rates. Such transient interactions may form but dissociate rapidly during wash steps, leading to a failure in detection [26].
SPR eliminates this risk by observing the interaction as it happens. This is particularly vital in therapeutic development. For instance, off-target binding of drugs, even if weak and transient, can lead to adverse drug reactions (ADRs) and is a major contributor to drug failures in clinical trials. It is estimated that approximately 75% of ADRs are due to dose-limiting toxicity, largely due to off-target interactions [26]. SPR-based secondary pharmacological profiling can reduce false-negatives, flagging problematic compounds earlier in the discovery pipeline [26].
Furthermore, kinetic rate constants provide deeper insight than affinity (KD) alone. The association rate (kₐ) and dissociation rate (kd) reveal the speed of complex formation and its stability, respectively. This is crucial for optimizing therapeutics where residence time (half-life of the bound complex) is critical for efficacy [26].
Benefits of a Label-Free Approach
The label-free nature of SPR offers several key advantages:
- Preservation of Native Activity: The absence of fluorescent or radioactive labels prevents potential alteration of the biomolecule's structure, conformation, or binding activity, ensuring data reflects true biological behavior [25].
- Reduced Assay Complexity: Eliminating the need for labeling streamlines experimental workflows, reduces preparation time, and avoids the costs associated with labeling reagents.
- Direct Observation: Binding is measured directly through mass change at the sensor surface, unlike indirect methods that rely on reporter signals which may be influenced by environmental factors or fail for certain molecules.
Experimental Protocols and Data Analysis
Detailed SPR Experimental Methodology
A representative SPR experiment, as utilized in recent studies, involves a series of carefully orchestrated steps to ensure high-quality, reproducible data [26].
Protocol: SPOC-Based SPR Screening
- Sensor Chip Functionalization: Biosensor chips are coated with a chloroalkane-linked surface to capture HaloTag fusion proteins.
- On-Chip Protein Synthesis (SPOC Method):
- Plasmid DNA containing HaloTag fusion protein open-reading frames is printed into nanowells of a specialized slide.
- This slide is affixed to a Protein NanoFactory system along with the functionalized biosensor chip.
- HeLa in vitro transcription and translation (IVTT) cell-free extract is injected over the nanowell slide.
- The system is incubated (e.g., 2 hours at 30°C) to allow cell-free protein synthesis and simultaneous covalent capture of the HaloTag fusion proteins directly onto the biosensor surface.
- The surfaces are subsequently rinsed with PBST (Phosphate-Buffered Saline with Tween-20) [26].
- Ligand Immobilization (Alternative Amine-Coupling): For traditional methods, a ligand (e.g., a protein) can be immobilized on a sensor chip via amine-coupling chemistry. This typically involves:
- Surface activation with a mixture of N-hydroxysuccinimide (NHS) and N-ethyl-N'-(dimethylaminopropyl)carbodiimide (EDC).
- Injection of the ligand to covalently bind to the activated esters.
- Blocking of remaining activated groups with ethanolamine [27].
- Binding Kinetics Measurement:
- A baseline is established by flowing running buffer (e.g., PBS) over the sensor surface.
- The analyte (e.g., an antibody or drug candidate) is injected over the surface at a defined concentration and flow rate (e.g., 0.5 μL/s [27]) for a set association phase period (e.g., 600 seconds).
- Running buffer is then flowed again to monitor the dissociation phase (e.g., 900 seconds).
- This process is repeated for a series of analyte concentrations to generate a full kinetic dataset [27].
- Surface Regeneration: The sensor surface is regenerated by injecting a solution that disrupts the binding interaction (e.g., 6 M Guanidine Hydrochloride, pH 1.5 [27]) without damaging the immobilized ligand, allowing for multiple analysis cycles on the same spot.
Data Evaluation and Kinetic Analysis
The analysis of SPR sensorgrams is a critical step. While commercial software packages exist, understanding the underlying principles is vital for correct interpretation [27]. The simplest model for one-to-one interaction is described by the following equations:
Association phase: ( dR/dt = ka \cdot C \cdot (R{max} - R) - kd \cdot R ) Dissociation phase: ( dR/dt = - kd \cdot R )
Where:
- ( R ) is the response at time ( t )
- ( R_{max} ) is the maximum binding capacity
- ( C ) is the analyte concentration
- ( k_a ) is the association rate constant (M⁻¹s⁻¹)
- ( k_d ) is the dissociation rate constant (s⁻¹)
The equilibrium dissociation constant is calculated as ( KD = kd / k_a ) (M).
Several mathematical approaches can be used to evaluate the binding curves, including linear transformations using derivatives or integrals, and non-linear fitting to the integrated rate equations [27]. It is essential to design experiments to avoid artifacts such as mass transport limitation (e.g., by using fast flow rates and low immobilization levels) and to verify that the chosen kinetic model adequately fits the experimental data [27].
Table 1: Key Kinetic Parameters Obtained from SPR Analysis
| Parameter | Symbol | Unit | Biological Significance |
|---|---|---|---|
| Association Rate Constant | kₐ | M⁻¹s⁻¹ | Speed of complex formation. |
| Dissociation Rate Constant | kd | s⁻¹ | Stability of the complex; speed of dissociation. |
| Equilibrium Dissociation Constant | K_D | M | Affinity; lower value indicates tighter binding. |
| Half-Life of Complex | t₁/₂ | s | Time for half of the complex to dissociate. |
Case Study: SPR vs. Endpoint Assay for Transient Interactions
A 2025 study provides a compelling case to illustrate the advantage of real-time SPR [26]. The research leveraged the SPOC platform to express HaloTag fusion proteins directly on SPR biosensors. Two different commercial antibodies (Ab #1 and Ab #2) targeting the HaloTag antigen were characterized.
- Endpoint Fluorescence Assay: When tested using a traditional immunofluorescent endpoint assay (involving multiple wash and incubation steps), the two antibodies showed disparate binding results. One antibody appeared to bind effectively, while the other showed markedly weaker signal, suggesting a potential false negative.
- Real-Time SPR Assay: In contrast, real-time SPR demonstrated that both antibodies were similarly capable of binding to the HaloTag proteins on the biosensor surface. The sensorgrams revealed that the antibody showing weak fluorescence had a fast dissociation rate (high kd). It bound to the antigen but dissociated so rapidly that it was washed away before detection in the endpoint assay.
This case underscores a key limitation of endpoint assays: their inability to reliably detect transient interactions characterized by fast kinetics. SPR's real-time monitoring eliminates this pitfall, providing a more accurate and comprehensive picture of the binding event [26].
Table 2: Comparison of Endpoint vs. Real-Time SPR Assays
| Feature | Traditional Endpoint Assay | Real-Time SPR |
|---|---|---|
| Detection Method | Indirect (e.g., fluorescence, radioactivity) | Direct, label-free |
| Time Resolution | Single measurement post-wash | Continuous, real-time monitoring |
| Kinetic Data | No | Yes (kₐ, kd) |
| Affinity Data | Indirect estimation | Direct calculation (K_D) |
| Risk of False Negatives | High for fast-dissociating complexes | Low |
| Information on Conformational Change | Limited | Possible |
| Throughput | Variable | High (e.g., SPOC allows ~864 spots [26]) |
The Scientist's Toolkit: Essential Research Reagents and Materials
Successful SPR experiments require careful selection of reagents and materials. The following table details key components used in the featured SPOC experiment and broader SPR practice [26].
Table 3: Key Research Reagent Solutions for SPR Experiments
| Reagent / Material | Function in the Experiment | Example from Literature |
|---|---|---|
| Functionalized SPR Chip | Solid support for immobilizing one interactant (ligand). | Chloroalkane-coated biosensor for HaloTag capture [26]. |
| Cell-Free Protein Synthesis System | Produces the protein ligand directly on-chip, enabling high-throughput library screening. | HeLa IVTT extract (e.g., from ThermoFisher) [26]. |
| Ligand (Immobilized) | The molecule immobilized on the sensor chip surface. | HaloTag fusion proteins expressed via SPOC [26]. |
| Analyte | The molecule in solution whose binding is measured. | Anti-HaloTag antibodies (e.g., from Proteintech, Promega) [26]. |
| Running Buffer | Liquid phase for analyte dilution and baseline stabilization. | Phosphate-Buffered Saline (PBS), often with surfactant (PBST) [26]. |
| Regeneration Solution | Removes bound analyte without damaging the ligand, enabling chip re-use. | Guanidine Hydrochloride (GdnHCl) solution, low pH buffers [27]. |
Visualizing the SPR Workflow and Advantage
The following diagrams, created using DOT language and adhering to the specified color and contrast guidelines, illustrate the core SPR workflow and its key advantage.
Diagram 1: Generic SPR Experimental Cycle
Diagram 2: SPR Avoids False Negatives from Transient Binding
Surface Plasmon Resonance stands as a powerful technique within the foundational toolkit of biochemical research due to its unique real-time and label-free capabilities. By enabling the direct observation of binding events as they occur, SPR provides robust, kinetic data that is essential for characterizing complex biomolecular interactions, especially those with fast dissociation rates that are prone to being missed by traditional methods. As demonstrated through contemporary experimental cases and protocols, the application of SPR in areas like off-target screening and therapeutic affinity optimization is invaluable for driving innovation in basic research and accelerating the development of safer, more effective biopharmaceuticals.
SPR in Practice: Methodologies and Applications in Drug Discovery and Diagnostics
Surface Plasmon Resonance (SPR) is a powerful optical technique that enables the label-free, real-time monitoring of biomolecular interactions. [28] [29] The fundamental principle involves the detection of changes in the refractive index at the surface of a sensor chip, which is typically coated with a thin gold film. [4] [30] When polarized light hits this metal film under specific conditions, it generates an evanescent wave that is exquisitely sensitive to changes in mass on the chip surface. [6] [1] As analytes bind to immobilized ligands, the increase in mass causes a proportional shift in the resonance angle or wavelength, which is measured in resonance units (RU) and recorded as a sensorgram. [4] [30] This setup provides researchers with detailed quantitative data on interaction kinetics, affinity, and specificity, making it indispensable in modern pharmaceutical research and drug discovery. [28]
The Heart of the System: SPR Sensor Chips
The sensor chip is often considered the "heart" of an SPR instrument. [28] Its surface must be meticulously designed to immobilize an adequate density of bio-recognition molecules while concurrently minimizing non-specific interactions, which is critical for the reliability and accuracy of the biosensor's performance. [28]
Covalent Coupling Sensor Chips
Covalent coupling chips allow for the direct and stable attachment of ligands to the sensor surface using various chemistries.
- CM5 (Standard Dextran Matrix): The CM5 sensor chip features a carboxymethylated dextran hydrogel that forms a flexible, three-dimensional matrix, extending 100-200 nanometers from the surface. [31] [32] This structure allows for high protein immobilization capacity (up to 50 ng/mm³) and is excellent for studying interactions where there is a large difference in molecular weight between the ligand and analyte. [32] It is a general-purpose chip suitable for all biomolecules, including small molecules, proteins, and nucleic acids. [31] [30]
- CM3 (Short Dextran): The CM3 chip provides the same chemistry as the CM5 but with a shorter dextran matrix. This is valuable for work with large analytes like cells and virus particles, as it can reduce steric hindrance. It may also give reduced non-specific binding when working with complex samples like serum. The immobilization yield is approximately 30% of that obtained on CM5. [31]
- CM4 (Low Carboxylation): The CM4 chip has a dextran matrix like CM5 but with a lower degree of carboxylation, making it less negatively charged. This surface is ideal for reducing non-specific binding of highly positively charged molecules or when working with crude samples like cell culture supernatants. Its immobilization yield is also about 30% of CM5's. [31]
- CM7 (High Carboxylation): The CM7 chip is comparable to CM5 but features a higher density of carboxymethylated dextran, giving it an approximately three times higher immobilization capacity. This surface is ideal for applications using small molecules and fragments but is generally less suitable for large biomolecular analytes. [31]
- Planar Sensor Chips: These chips feature a low-capacity, two-dimensional surface. One common type uses a mixed self-assembled monolayer (SAM) of alkanethiolates, combining polyethylene glycol-terminated (90%) and COOH-terminated (10%) thiols. The PEG chains minimize non-specific binding, while the COOH groups provide functional attachment sites. These surfaces are particularly useful for studying protein-protein interactions where a dextran matrix is undesirable. [32]
Capture-Based Sensor Chips
Capture chips utilize high-affinity interactions to immobilize ligands in a defined orientation, which can enhance binding activity and consistency.
- SA (Streptavidin Chip): The SA sensor chip is pre-immobilized with streptavidin for capturing biotinylated ligands. [33] The streptavidin-biotin interaction is one of the strongest non-covalent bonds known (KD ~ 10⁻¹⁵ M), offering high stability and reproducibility. [33] [34] This chip is a general-purpose tool for capturing biotinylated proteins, peptides, and nucleic acids, and it allows for the use of harsh regeneration conditions. [33] [32] The oriented capture often results in a more active surface compared to random covalent immobilization. [32]
- NTA (Nitrilotriacetic Acid Chip): The NTA sensor chip is functionalized with NTA for capturing histidine-tagged molecules, such as recombinant proteins. [30] The capture is mediated via Ni²⁺/NTA chelation, which provides a well-oriented attachment through the histidine tag. The surface is easily regenerated with an injection of imidazole or EDTA. [32] This chip is ideal for studying membrane proteins or any protein that can be expressed with a His-tag. [30]
- Protein A Chip: This chip is pre-immobilized with Protein A, which contains high-affinity binding sites for the Fc region of immunoglobulins from various species, including human and rabbit. It is specifically designed for the oriented capture of antibodies, leaving their antigen-binding sites free. Optimal binding occurs at pH 8.2. [32]
Specialized Sensor Chips
- Hydrophobic Sensor Chips (e.g., HPA, L1): These chips are designed for lipid-based studies. The HPA chip presents a flat hydrophobic surface for the formation of lipid monolayers, useful for studying membrane-binding biomolecules. [30] The L1 chip is a dextran matrix modified with lipophilic substances, enabling the capture of intact liposomes and retention of the lipid bilayer structure. [30]
- Plain Gold Sensor Chips: A bare gold surface is useful for studying adsorption in real-time, applying user-defined coating chemistries, or for experiments combining SPR with electrochemistry. [32]
Table 1: Summary of Key SPR Sensor Chips and Their Characteristics
| Sensor Chip | Surface Chemistry | Immobilization Method | Key Characteristics | Ideal Applications |
|---|---|---|---|---|
| CM5 [31] [30] | Carboxymethylated dextran | Covalent (Amine, Thiol, etc.) | High capacity; 3D hydrogel; versatile | General-purpose; wide range of molecular weights |
| CM3 [31] | Short carboxymethylated dextran | Covalent | Short matrix; reduced steric hindrance | Large analytes (cells, viruses); reduced NSB |
| CM4 [31] | Low-carboxylation dextran | Covalent | Less charged; reduced NSB | Positively charged molecules; crude samples |
| SA [33] [32] | Streptavidin on dextran | Affinity Capture (Biotin) | High-affinity, oriented capture | Biotinylated proteins, peptides, DNA |
| NTA [32] [30] | NTA on dextran | Affinity Capture (His-tag) | Oriented capture; metal chelation | His-tagged recombinant proteins |
| Protein A [32] | Protein A on dextran | Affinity Capture (Fc region) | Specific antibody capture | Antibody-antigen interactions |
| Planar [32] | SAM with PEG and COOH | Covalent | Low capacity; minimal NSB | Protein-protein interactions |
Experimental Protocol: A Step-by-Step Guide
A typical SPR experiment follows a logical workflow to ensure the collection of high-quality, reproducible data. The process can be broken down into several key phases, as illustrated below.
Surface Preparation and Ligand Immobilization
The first critical step is to attach the ligand to the sensor chip surface in a stable and functional manner.
- Chip Selection: Choose the appropriate sensor chip based on the nature of the ligand and analyte, as well as the experimental goals (see Table 1). [32] [30]
- Surface Activation (For Covalent Chips): For CM-series chips, the carboxyl groups on the dextran are typically activated using a mixture of N-ethyl-N'-(dimethylaminopropyl)carbodiimide (EDC) and N-hydroxysuccinimide (NHS). This creates an amine-reactive ester for coupling. [33]
- Ligand Immobilization:
- For covalent coupling, the ligand is dissolved in a low-ionic strength buffer at a pH below its isoelectric point to ensure a positive charge, facilitating electrostatic pre-concentration onto the negatively charged dextran surface. It is then injected over the activated surface. [32]
- For capture coupling, the biotinylated or His-tagged ligand is injected in a suitable running buffer or a buffer containing salt (e.g., 0.5 M NaCl for long nucleic acid fragments) to promote binding to the SA or NTA surface. [33] [32]
- Surface Blocking (For Covalent Chips): After ligand attachment, any remaining activated esters are deactivated by injecting ethanolamine. [33]
Binding Interaction and Regeneration
Once the ligand is immobilized, the binding analysis with the analyte can begin.
- Analyte Injection: A series of analyte concentrations, prepared in running buffer, are injected over the ligand surface and a reference surface at a constant flow rate. The association phase is monitored in real-time as the analyte binds. [4] [1] The concentration range should ideally span from 10-fold below to 10-fold above the expected KD. [30]
- Dissociation Phase: The injection is stopped, and running buffer is flowed over the surface. The decrease in signal as the analyte dissociates is monitored. [1] [30]
- Surface Regeneration: To prepare the surface for the next analyte injection, a regeneration solution is injected to disrupt the ligand-analyte interaction without denaturing the immobilized ligand. [33] [1] Common regeneration agents include:
Table 2: Common Regeneration Solutions and Their Applications
| Regeneration Solution | Typical Use Cases | Considerations |
|---|---|---|
| Low pH (e.g., 10 mM Glycine, pH 2.0) [33] | Antibody-antigen complexes | Check ligand stability in acidic conditions |
| High pH (e.g., 50 mM NaOH) [33] | Protein-protein interactions; DNA hybrids | Can be too harsh for some proteins |
| High Salt (e.g., 2-4 M NaCl, 3 M MgCl₂) [33] | Electrostatic interactions | Mild option for sensitive ligands |
| 10-100 mM Imidazole [32] | NTA chip regeneration | Competes with His-tag for NTA binding |
| 10-350 mM EDTA [32] | NTA chip regeneration | Chelates and removes Ni²⁺ ions |
The Scientist's Toolkit: Essential Reagents and Materials
Successful SPR experiments require careful preparation and the use of specific, high-quality materials. The following table lists key reagents and their functions.
Table 3: Essential Research Reagent Solutions for SPR
| Item | Function / Purpose | Key Details |
|---|---|---|
| Sensor Chips [4] [32] | Platform for ligand immobilization and binding detection | Choice depends on ligand properties and assay goal (see Table 1). |
| Running Buffer [4] | Liquid medium for sample dilution and continuous flow | Must include detergent (e.g., 0.05% Tween 20) to minimize NSB. Buffer, salt, and DMSO concentrations must be fixed. |
| EDC & NHS [33] | Activating agents for carboxylated surfaces (e.g., CM-series) | Forms amine-reactive esters for covalent ligand coupling. |
| Ethanolamine [33] | Blocking agent | Deactivates remaining esters after covalent immobilization. |
| Regeneration Solutions [33] [32] | Removes bound analyte to regenerate the ligand surface | Must be strong enough to dissociate the complex but not damage the ligand (see Table 2). |
| Instrument Cleaning Solutions [4] | Maintains instrument and fluidic path hygiene | Includes solutions like Desorb 1, Desorb 2, and Biadisinfectant. |
The selection of an appropriate sensor chip and a well-optimized experimental protocol are foundational to successful SPR research. From the versatile CM5 to the oriented capture offered by SA and NTA chips, each surface provides unique advantages tailored to specific biological questions. Mastery of surface chemistry, immobilization strategies, and regeneration protocols allows researchers to obtain highly reliable kinetic and affinity data. As SPR technology continues to evolve, integration with advanced materials and data analysis techniques will further solidify its role as a critical tool in drug discovery and biochemical research. [28]
In Surface Plasmon Resonance (SPR) research, the immobilization of a ligand onto a sensor surface is a critical foundational step. The method of attachment is not merely a technicality but a decisive factor that can influence the activity of the ligand, the accessibility of its binding site, and the ultimate accuracy of the kinetic and affinity data derived from the experiment [35] [36]. The core challenge lies in creating a stable, functional, and homogeneous layer of binding sites without altering the native properties of the immobilized molecule. Within this context, two principal philosophies have emerged: covalent coupling and capture methods. This guide provides an in-depth technical comparison of these strategies, detailing their mechanisms, applications, and experimental protocols to inform best practices in fundamental SPR research and drug development.
Core Principles of SPR Immobilization
The fundamental requirement of any SPR immobilization strategy is to permanently or stably attach one interactant (the ligand) to the sensor surface while preserving its biological activity and allowing unhindered access to its binding partner (the analyte) in solution [36]. The ideal immobilization achieves several goals:
- Preserved Activity: The attachment should not occur in or sterically block the active site of the ligand.
- Minimal Heterogeneity: The ensemble of immobilized molecules should be as uniform as possible to be well-described by a single set of binding parameters. Inhomogeneous attachment can lead to a dispersion of binding energies and complex, difficult-to-interpret data [35].
- Stability: The ligand should remain attached throughout the binding and regeneration cycles of the experiment.
- Orientation: When possible, the ligand should be oriented such that its binding site is freely accessible to the analyte in solution.
Failure to adequately address these aspects can introduce artifacts, such as non-specific binding, altered kinetics, or a significant fraction of inactive ligand, compromising the validity of the interaction data [35].
Covalent Coupling Immobilization
Covalent coupling involves forming irreversible chemical bonds between functional groups on the ligand and reactive groups on the sensor surface. This method is widely applicable and provides a stable surface that can withstand multiple regeneration cycles [37].
Mechanism and Common Chemistries
The most prevalent covalent coupling chemistry utilizes EDC/NHS to activate carboxyl groups on a sensor chip surface (e.g., Carboxyl sensors), which then react with primary amine groups (e.g., on lysine residues) on the ligand [37] [38]. Alternative chemistries allow for coupling via other functional groups:
- Amine Sensors: The surface contains amine groups, which are used to couple to carboxyl groups on the ligand after EDC/NHS activation. This is less common for proteins but can be useful for ligands with carboxylic acid tags [37].
- Thiol Coupling: Surfaces can be activated to react with free thiol groups (-SH) on the ligand, such as those on cysteine residues. This can offer more controlled orientation if a unique cysteine is available [38].
- Aldehyde Coupling: Activated surfaces can form bonds with aldehyde groups, which can be useful for specifically immobilizing glycoproteins [38].
Experimental Protocol: Amine Coupling via EDC/NHS
The following table outlines a standardized protocol for amine coupling on a carboxyl sensor surface, as utilized in various SPR systems [38].
Table 1: Standardized Experimental Protocol for Amine Coupling
| Step | Reagent | Purpose | Duration (Example) | Flow Rate (Example) |
|---|---|---|---|---|
| 1. Activation | EDC/NHS mixture | Activates carboxyl groups on the sensor surface, forming NHS esters. | 6-7 minutes | 5-10 µL/min [38] |
| 2. Immobilization | Ligand in low-salt buffer (e.g., pH 5.5 acetate) | Ligand's primary amines react with NHS esters, forming covalent amide bonds. | Controlled to achieve desired density | 5-10 µL/min [38] |
| 3. Blocking | Ethanolamine-HCl | Deactivates any remaining NHS esters on the surface to prevent non-specific binding. | 6-7 minutes | 5-10 µL/min [38] |
Advantages and Limitations
Advantages:
- High Stability: The covalent bond minimizes ligand dissociation, maintaining binding capacity over long experiments [37].
- Broad Applicability: Compatible with most biomolecules that possess amine, carboxyl, or thiol groups, minimizing the need for genetic modification [37].
- Regeneration Tolerance: The stable surface can often withstand harsh regeneration conditions to remove tightly bound analyte.
Limitations:
- Random Orientation: Coupling via common amino acids (e.g., lysines) leads to a heterogeneous population of ligands with varying orientations, which can block the active site for a subset of molecules [35] [37].
- Potential Activity Loss: The chemical reaction can, in some cases, alter the ligand's conformation or directly modify residues critical for binding [36].
- Surface-Induced Heterogeneity: The proximity to the sensor surface and its rugose environment can create microenvironments that affect the binding energetics of the immobilized ligand [35].
Capture Immobilization
Capture methods immobilize the ligand through a non-covalent, high-affinity interaction with a molecule that is itself covalently attached to the sensor surface. This approach often provides superior control over ligand orientation and activity.
Mechanism and Common Systems
- Antibody-Antigen Capture: A highly specific antibody is first covalently immobilized. This antibody then captures the ligand of interest from a solution, ensuring a defined orientation [37]. For instance, an anti-His antibody can be used to capture His-tagged ligands [38].
- Streptavidin-Biotin: One of the strongest non-covalent interactions known (KD ~10-15 M). Surfaces are coated with streptavidin (or neutravidin) to capture biotinylated ligands. This method offers near-irreversible attachment and excellent orientation control [39] [37] [38].
- Protein A/G for Antibodies: Protein A or G is immobilized on the surface, which specifically binds the Fc region of IgG antibodies. This orientates antibody ligands with their antigen-binding sites facing the solution, maximizing their activity [37] [38].
- NTA for His-Tagged Proteins: A nitrilotriacetic acid (NTA) surface, charged with nickel ions (Ni2+), captures proteins containing a polyhistidine tag (e.g., His6). While convenient, this method can suffer from slow dissociation of the ligand and metal-ion-induced idiosyncrasies [39].
Experimental Protocol: Capture Coupling for His-Tagged Proteins
A robust "capture-coupling" method combines the orientation benefits of capture with the permanence of covalent attachment. The following workflow details this procedure for a His-tagged ligand [39].
Diagram: The Capture-Coupling Workflow for His-Tagged Proteins
Table 2: Detailed Steps for His-Tag Capture-Coupling
| Step | Reagent / Action | Purpose |
|---|---|---|
| 1. Surface Preparation | Dock NTA sensor chip and prime system with appropriate buffers. | Prepares the SPR instrument and fluidics for immobilization [39]. |
| 2. Nickel Charging | Inject a solution of NiSO4 (e.g., 500 µM). | Loads the NTA surface with Ni2+ ions, enabling His-tag binding [39]. |
| 3. Ligand Capture | Inject the His6-tagged fusion protein at a slow flow rate (e.g., 5 µL/min). | Captures the ligand onto the surface in a specific orientation via its His-tag [39]. |
| 4. Covalent Stabilization | Inject EDC/NHS coupling solution. | Activates carboxyl groups on the captured ligand or the dextran matrix. |
| 5. Covalent Coupling | Inject the His6-tagged ligand a second time. | The newly activated groups form covalent bonds with amine groups on the already-captured ligand, permanently fixing it to the surface [39]. |
| 6. Blocking & Regeneration | Inject ethanolamine to block, then EDTA to strip Ni2+. | Quenches unreacted groups and removes nickel, leaving a stable, covalently attached ligand layer [39]. |
Advantages and Limitations
Advantages:
- Controlled Orientation: The ligand is presented in a uniform, often optimal, orientation, which can maximize the fraction of active molecules [39] [37].
- Preserved Activity: By avoiding direct chemical modification of the ligand, its native conformation and binding function are more likely to be retained [37].
- Reusable Surface: The capture molecule (e.g., Protein A, NTA) can often be regenerated by stripping off the old ligand and capturing a fresh one, making the method efficient and cost-effective for screening [37] [38].
- Suitable for Complex Samples: Allows for the specific capture of a ligand from a crude mixture, such as cell lysates [37].
Limitations:
- Ligand Dissociation: Some capture systems (notably NTA) can exhibit slow but continuous dissociation of the ligand, which can complicate data analysis [39].
- Capacity Limitation: The binding capacity is dependent on the density of the capture molecule.
- Requires Modification: The ligand must possess or be engineered to have a specific tag (e.g., His, biotin), which may not always be feasible [37].
Strategic Comparison and Selection Guide
Choosing between covalent and capture methods depends on the experimental goals, the nature of the ligand, and the required data quality. The following diagram and table provide a direct comparison to guide this decision.
Diagram: Decision Workflow for Immobilization Strategy
Table 3: Direct Comparison of Covalent Coupling vs. Capture Methods
| Parameter | Covalent Coupling | Capture Methods |
|---|---|---|
| Stability | High; irreversible attachment [37]. | Variable; can range from high (biotin-streptavidin) to moderate (NTA) [39] [37]. |
| Orientation | Random; can lead to heterogeneity and inactive sites [35] [37]. | Directed; promotes a uniform, active population of ligands [39] [37]. |
| Ligand Requirement | Requires accessible functional groups (amine, thiol, carboxyl). | Requires a specific tag or epitope (biotin, His-tag, Fc region) [37]. |
| Surface Reusability | Low; ligand is permanently attached. | High; capture surface can often be regenerated and reused for a new ligand [37] [38]. |
| Risk of Altered Activity | Higher; chemical reaction may affect the binding site [36]. | Lower; gentle, specific capture helps preserve native conformation [37]. |
| Ideal Use Case | Stable, robust surfaces for long-term studies; ligands without tags. | Screening applications; sensitive ligands where orientation is key; tagged proteins [39] [37]. |
The Scientist's Toolkit: Essential Reagents and Materials
Successful SPR immobilization requires a suite of specialized reagents and sensor chips. The following table catalogues key solutions for the researcher's toolkit.
Table 4: Research Reagent Solutions for SPR Immobilization
| Reagent / Material | Function | Example Use Cases |
|---|---|---|
| Carboxyl Sensor Chips | The standard surface for EDC/NHS-based amine coupling of proteins and other biomolecules [37]. | General covalent immobilization of ligands via lysine residues. |
| NTA Sensor Chips | For capturing His-tagged proteins via chelated Ni2+ ions [39] [38]. | Capture and study of recombinant His-tagged proteins; can be used in capture-coupling protocols. |
| Streptavidin/Biotin Sensor Chips | For capturing biotinylated ligands with very high affinity [37] [38]. | Immobilization of biotinylated DNA, proteins, or other molecules where orientation and stability are paramount. |
| Protein A/G Sensor Chips | For capturing a broad range of IgG antibodies via their Fc region [37] [38]. | Oriented immobilization of antibody ligands for antigen-binding studies. |
| EDC/NHS Coupling Kit | Contains the chemicals (EDC and NHS) required to activate carboxylated surfaces for covalent coupling [39] [38]. | Essential for amine coupling on carboxyl sensors and for the capture-coupling method. |
| Regeneration Buffers | Solutions (e.g., low pH glycine, EDTA) used to break the analyte-ligand bond without damaging the immobilized ligand [38]. | Restoring the ligand surface between analyte injections in a kinetic experiment. |
The selection between covalent coupling and capture strategies is a fundamental decision in the design of any SPR experiment. Covalent coupling offers simplicity and robust stability, making it a versatile, widely applicable choice. In contrast, capture methods excel in providing controlled orientation and preserving ligand activity, which is often critical for obtaining accurate kinetic data, particularly for therapeutic antibody characterization or tagged recombinant proteins. As SPR continues to be a cornerstone technique in drug discovery and basic research—evidenced by its growing market [40]—the strategic optimization of immobilization remains paramount. Researchers must weigh the trade-offs of each method, considering their specific ligand and experimental needs. Techniques like affinity distribution analysis [35] and hybrid methods like capture-coupling [39] provide powerful means to assess and ensure the quality of the prepared sensor surface, ultimately leading to more reliable and insightful biomolecular interaction data.
Within the framework of Surface Plasmon Resonance (SPR) research fundamentals, running buffer optimization is a critical prerequisite for generating robust and reliable biosensor data. A well-composed and properly prepared running buffer ensures baseline stability, minimizes non-specific interactions, and provides an optimal environment for biomolecular binding. This technical guide provides an in-depth examination of running buffer composition, essential additives, and preparation protocols, with a focused emphasis on degassing procedures to counteract baseline drift and artifacts. Adherence to these methodologies is essential for researchers and drug development professionals aiming to obtain publication-quality kinetic and affinity data.
In Surface Plasmon Resonance, the running buffer is the liquid phase that continuously flows over the sensor chip surface. Its primary functions are to:
- Maintain a Stable Baseline: The buffer's refractive index must be constant, as the SPR signal is exquisitely sensitive to changes in the refractive index of the medium near the sensor surface. Fluctuations cause signal drift and noise, obscuring genuine binding events [41] [4].
- Provide a Physiological Environment: The buffer must preserve the stability, solubility, and biological activity of both the immobilized ligand and the injected analyte throughout the experiment.
- Minimize Unwanted Interactions: Through optimal pH, ionic strength, and specific additives, the running buffer suppresses non-specific binding (NSB) of the analyte to the sensor chip or the ligand matrix [42].
Failure to optimize the running buffer is a primary source of experimental artifacts, including bulk shifts, high non-specific binding, and irreversible complex formation, which can compromise data integrity and lead to erroneous conclusions in drug discovery pipelines [16].
Core Buffer Composition and Selection
The choice of a base buffer is the foundation of any SPR experiment. The selected buffer must have good buffering capacity at the desired pH and be compatible with the biomolecules under investigation.
Table 1: Common Base Buffers for SPR Experiments
| Buffer | Typical pH Range | Key Advantages | Considerations for Use |
|---|---|---|---|
| HEPES | 7.0 - 8.0 | Chemically stable; non-reactive; common in biochemical assays. | Does not contain metal ions; may require supplementation. |
| Phosphate Buffered Saline (PBS) | 7.2 - 7.4 | Physiologically relevant; widely used. | Prone to microbial growth; phosphate can interfere with some coupling chemistries. |
| Tris-HCl | 7.0 - 9.0 | Effective in a broad alkaline range. | Sensitive to temperature; can act as a nucleophile in certain reactions. |
| Acetate | 4.0 - 5.5 | Ideal for low pH immobilization of ligands via amine coupling. | Low buffering capacity at neutral pH. |
| Borate | 8.0 - 10.0 | Suitable for high pH immobilization or binding studies. | Less common for standard physiological conditions. |
Key Chemical Parameters
- pH: The pH of the running buffer significantly influences the charge and three-dimensional structure of proteins, thereby affecting their binding affinity and kinetics. It is critical to use a pH that maintains ligand and analyte stability, typically near their isoelectric point to reduce non-specific charge-based interactions [42].
- Ionic Strength: The salt concentration (e.g., NaCl) in the buffer shields charged groups on proteins and the sensor surface. An appropriate ionic strength is crucial; concentrations that are too low can promote electrostatic non-specific binding, while concentrations that are too high may disrupt hydrophobic interactions or even precipitate proteins [42].
Essential Additives and Their Functions
Additives are incorporated into the running buffer to enhance specificity, stabilize biomolecules, and prevent surface fouling. The running buffer used during analyte injection must exactly match the buffer in which the analyte is dissolved to prevent bulk shift effects [42].
Table 2: Common Running Buffer Additives and Their Applications
| Additive | Typical Concentration | Primary Function | Mechanism of Action & Notes |
|---|---|---|---|
| Tween 20 (or other non-ionic surfactants) | 0.005% - 0.05% | Reduce hydrophobic non-specific binding (NSB) [42]. | Disrupts hydrophobic interactions between the analyte and sensor surface. Higher concentrations may denature proteins. |
| Bovine Serum Albumin (BSA) | 0.1 - 1.0 mg/mL | Block non-specific protein-binding sites [42]. | A globular protein that coats surfaces, shielding molecules from NSB. Critical Note: BSA should be added to the analyte sample buffer only, not during ligand immobilization, to prevent blocking the ligand. |
| Salts (e.g., NaCl) | 150 mM - 1 M | Reduce charge-based NSB and modulate binding affinity [42]. | Shields charged groups on proteins and the dextran matrix. Concentration requires optimization. |
| Chelators (e.g., EDTA) | 1 - 10 mM | Prevent metal-ion catalyzed protein aggregation or degradation. | Chelates divalent cations; essential for metal-sensitive proteins. |
| DMSO | ≤ 1-3% (v/v) | Solubilize small molecule analytes. | A common source of bulk refractive index shift; concentration must be matched perfectly between all analyte samples and the running buffer. |
The Critical Role of Degassing and Protocol
Degassing the running buffer is a non-negotiable step in SPR preparation. Dissolved air in the buffer can form microscopic bubbles when the buffer passes through the flow system and is warmed by the instrument. These bubbles cause severe signal spikes and baseline instability because air has a dramatically different refractive index than the liquid [41].
Detailed Degassing Methodology
Principle: Reducing the pressure above the buffer lowers the solubility of gases, causing them to escape from the liquid.
Materials:
- Running buffer solution
- Vacuum degassing system (or ultrasonic bath)
- Vacuum flask and trap
- Sterile, disposable 0.22 µm filter unit
Step-by-Step Protocol:
- Preparation: Filter the buffer through a 0.22 µm filter into a clean, scrupulously rinsed vacuum flask. Filtration removes particulate matter that can nucleate bubble formation.
- Degassing: Place the flask on a magnetic stirrer and connect it to a vacuum source. Begin stirring vigorously. Apply a gentle vacuum for 20-30 minutes. The process is complete when no more bubbles are visible rising to the surface.
- Handling: After degassing, carefully release the vacuum. The buffer should be used immediately or stored in a sealed container to prevent re-equilibration with atmospheric gases.
- Verification: A properly degassed buffer will result in a smooth, stable baseline during the instrument's prime and equilibration steps, with no sudden, sharp "spikes" in the sensorgram.
The following workflow diagram summarizes the key steps and decision points in the buffer optimization process:
Experimental Protocol: A Systematic Approach to Optimization
This protocol outlines a step-wise method for empirically determining the optimal running buffer conditions for a novel molecular interaction.
Materials and Equipment
- SPR instrument (e.g., Biacore T200) [4]
- Sensor chips (e.g., CM5 series S)
- Ligand and analyte molecules
- Buffer components (as listed in Tables 1 & 2)
- pH meter and analytical balance
- Vacuum degassing system
- 0.22 µm sterile filters
Scouting and Optimization Procedure
Preliminary Scouting:
- Immobilize a small amount of ligand on a sensor chip using a standard coupling method.
- Prepare a series of running buffers with varying pH (e.g., 6.0, 7.4, 8.0) and ionic strengths.
- Inject a single, medium concentration of analyte in each buffer. Observe the response level, shape of the sensorgram, and the baseline stability.
Non-Specific Binding (NSB) Test:
- Once a base buffer is selected, test for NSB by injecting a high concentration of analyte over a reference flow cell with no immobilized ligand [42].
- If a significant response is observed (>10 RU), systematically introduce additives from Table 2 (e.g., 0.05% Tween 20, then 1 mg/mL BSA in analyte buffer only) and repeat the NSB test until the signal is minimized.
Bulk Shift Assessment:
- Prepare the final analyte dilution series in the optimized running buffer.
- In the raw sensorgram data (before reference subtraction), inspect the start and end of each analyte injection for a "square" shaped response. This indicates a bulk refractive index shift [42].
- Ensure the running buffer and analyte sample buffer are perfectly matched. If a bulk shift persists from a necessary additive like DMSO, confirm that the instrument's reference subtraction function adequately corrects for it.
The Scientist's Toolkit: Key Research Reagent Solutions
Table 3: Essential Materials for SPR Running Buffer and Assay Preparation
| Reagent / Material | Function in SPR | Application Notes |
|---|---|---|
| HEPES Buffer | Provides a stable, non-reactive pH environment for biomolecular interactions. | Preferred over phosphate buffers for carbodiimide coupling chemistry. |
| Tween 20 | Non-ionic surfactant that coats surfaces to minimize hydrophobic non-specific binding. | Typically used at 0.05% (v/v); higher concentrations risk protein denaturation. |
| BSA (Fatty-Acid Free) | Carrier protein used to block non-specific binding sites on proteins and surfaces. | Use in analyte sample buffer only; fatty-acid free grade is recommended. |
| DMSO (Ultra-Pure Grade) | Universal solvent for small molecule analytes; prevents compound precipitation. | Concentration must be strictly matched (e.g., ±0.1%) in all samples and running buffer to prevent bulk shift [42]. |
| Sensor Chips (e.g., CM5, NTA) | The solid support with a gold film and modified matrix for ligand immobilization. | Choice depends on ligand properties and coupling chemistry (e.g., CM5 for amine coupling) [42] [4]. |
| Desorb Solution (e.g., Desorb 1, Desorb 2) | Instrument cleaning solutions for rigorous maintenance of the fluidic system. | Used according to a scheduled protocol to prevent carryover and system contamination [4]. |
Running buffer optimization is a foundational element of SPR experimental design that directly dictates data quality and interpretability. A methodical approach involving the selection of a physiologically relevant base buffer, the strategic inclusion of additives like surfactants and carriers to mitigate NSB, and the rigorous implementation of degassing protocols is paramount. By adhering to the detailed guidelines and protocols presented herein, researchers can significantly enhance baseline stability, minimize artifacts, and ensure that the obtained kinetic and affinity constants accurately reflect the true biology of the molecular interaction under investigation.
Surface Plasmon Resonance (SPR) is a label-free biophysical technique used to study molecular interactions in real-time. It is widely employed to determine the specificity, affinity, and kinetics of binding events between a ligand and an analyte. A significant advantage of SPR is that it allows researchers to monitor binding events as they happen, providing direct measurement of association (kₐ) and dissociation (kᵈ) rate constants. These kinetic parameters are crucial in fields like drug development, as they reveal not just the strength of an interaction (affinity), but also the speed at which complexes form and break apart. The technique is highly sensitive, capable of detecting picomolar to nanomolar amounts of analyte and measuring interactions without the need for fluorescent or radioactive labeling [43]. This guide details the core principles and methodologies for determining kₐ and kᵈ within the context of fundamental SPR research.
Theoretical Foundations of Kinetic Analysis
The Langmuir Binding Model
For a simple 1:1 interaction between an analyte (A) and an immobilized ligand (B), the binding model is represented by: A + B ⇌ AB The association rate (kₐ) and dissociation rate (kᵈ) are defined by the differential rate equation: dR/dt = kₐ * C * (Rmax - R) - kᵈ * R where dR/dt is the rate of change of the SPR response, C is the analyte concentration, R is the response at time t, and Rmax is the maximum binding capacity of the surface [44].
Determining the Dissociation Rate Constant (kᵈ)
During the dissociation phase, the analyte in the flow is replaced by buffer (C=0). The equation simplifies to: dR/dt = -kᵈ * R Integration of this equation yields an expression that allows kᵈ to be determined from the exponential decay of the signal: R_t = R₀ * e^(-kᵈ * t) where R₀ is the response at the start of the dissociation phase. The dissociation rate constant (kᵈ) is therefore obtained by fitting the dissociation data to a single exponential decay [44]. The half-life of the complex can be calculated as ln(2)/kᵈ [44].
Determining the Association Rate Constant (kₐ)
The association phase is governed by both kₐ and kᵈ. The observed association rate (kₒbₛ) at a given analyte concentration (C) is: kₒbₛ = kₐ * C + kᵈ By measuring kₒbₛ at several different analyte concentrations and plotting kₒbₛ versus C, a linear fit yields kₐ from the slope and kᵈ from the y-intercept [44].
From Kinetics to Affinity
The equilibrium dissociation constant (KD), which describes the binding affinity, is the ratio of the kinetic rate constants: KD = kᵈ / kₐ A low K_D value indicates high affinity, which can result from a fast association (high kₐ), a slow dissociation (low kᵈ), or a combination of both [43].
Table 1: Interpretation of Kinetic Rate Constants and Affinity
| Parameter | Definition | High Value Indicates | Low Value Indicates |
|---|---|---|---|
| Association Rate (kₐ) | Speed of complex formation | Rapid binding | Slow binding |
| Dissociation Rate (kᵈ) | Speed of complex breakdown | Unstable complex, short residence time | Stable complex, long residence time |
| Affinity (K_D = kᵈ/kₐ) | Equilibrium dissociation constant | Weak binding (High K_D) | Strong binding (Low K_D) |
Table 2: Practical Implications of kᵈ Values on Complex Half-Life [44]
| kᵈ (s⁻¹) | Complex Half-Life (Seconds) | Complex Half-Life (Minutes) | Practical Consideration |
|---|---|---|---|
| 10⁻¹ | 7 | 0.1 | Very rapid dissociation |
| 10⁻² | 69 | 1.2 | Short dissociation |
| 10⁻³ | 693 | 11.6 | Moderately stable complex |
| 10⁻⁴ | 6,931 | 115.6 | Long-lived complex; requires extended dissociation monitoring |
Experimental Workflow for Kinetic Analysis
The following diagram illustrates the key stages of a kinetic experiment, from surface preparation to data analysis.
Sensor Surface Preparation
The first step involves immobilizing the ligand on a sensor chip. Various chip surfaces (e.g., carboxymethyl dextran CM5, lipophilic L1) are available to accommodate different types of ligands, from proteins to lipids. The immobilization must be performed to ensure ligand stability and activity throughout the experiment. For kinetic studies, a low ligand density is often preferred to minimize mass transport effects and rebinding artifacts [43].
Association and Dissociation Phases
As shown in the workflow, the experiment cycles through key phases. During the association phase, analyte is flowed over the ligand surface, and binding is monitored in real-time as an increase in the SPR signal (Response Units, RU). This is followed by the dissociation phase, where buffer is flowed over the surface, and the decrease in signal from the dissociation of the complex is recorded. To obtain reliable kinetic data, the dissociation should be monitored for a sufficient duration; as a rule of thumb, the curve should decrease by at least 5% before analysis is attempted [44]. For complexes with very slow dissociation (kᵈ < 10⁻⁴ s⁻¹), this may require dissociation times of an hour or more.
Data Analysis
The reference cell signal (a cell with no ligand or an irrelevant ligand) is subtracted from the active cell signal to account for bulk refractive index changes and non-specific binding. The resulting sensorgram data for multiple analyte concentrations are then fitted globally to the 1:1 Langmuir binding model to extract kₐ and kᵈ [44] [43]. The quality of the fit is assessed, and deviations may indicate a more complex interaction, such as heterogeneity in the ligand or analyte, or rebinding events [44].
The Scientist's Toolkit: Essential Reagents and Materials
Successful execution of a kinetic experiment requires careful preparation and the right materials. The following table lists key reagents and their functions.
Table 3: Essential Research Reagent Solutions for SPR Kinetics
| Reagent/Material | Function and Importance | Example / Key Consideration |
|---|---|---|
| Running Buffer | The liquid phase that carries the analyte; defines the chemical environment for the interaction. | Must match analyte storage buffer to minimize refractive index shifts. Should be degassed and detergent-free for system stability [43]. |
| Sensor Chip | The platform for ligand immobilization. The surface chemistry must be compatible with the ligand. | L1 Chip: For capturing liposomes or membrane proteins [43]. CM5 Chip: General-purpose dextran surface for covalent coupling. SA Chip: For capturing biotinylated ligands. |
| Ligand | The molecule immobilized on the sensor chip. | Should be highly pure and stable. Small tags (e.g., His-tag) are preferred over large tags that may interfere [43]. |
| Analyte | The molecule in solution that binds to the ligand. | Should be prepared in running buffer. If stored in glycerol, the running buffer should contain 5% glycerol to match refractive index [43]. |
| Regeneration Solution | A solution that removes bound analyte without damaging the immobilized ligand. | Allows for re-use of the sensor chip. Must be empirically determined for each interaction (e.g., 10-100 mM HCl, 10 mM Glycine pH 2.0-3.0, or mild detergents). |
| System Cleaning Solutions | Maintains instrument performance by removing contaminants from the fluidics. | Includes desorb solutions (e.g., 0.5% SDS) and sanitize solutions (e.g., 10% bleach). Run regularly with a maintenance chip [43]. |
Advanced Application: Measuring Ternary Complex Kinetics
SPR's application extends beyond simple 1:1 interactions. It can be used to study complex mechanisms, such as the formation of ternary complexes induced by bifunctional molecules like PROTACs (Proteolysis-Targeting Chimeras). The following diagram outlines the key steps and binding equilibria involved in a ternary complex kinetic assay.
In this assay, one protein (e.g., the E3 ligase VHL) is immobilized. The kinetics of the PROTAC binding to VHL alone are first measured to determine the binary affinity (KDbinary). Subsequently, the PROTAC is pre-incubated with a near-saturating concentration of the target protein, and this pre-formed complex is injected over the immobilized VHL to measure the ternary affinity (KDternary). The difference between these two values quantifies the cooperativity (α) of the interaction. A key finding from such studies is that the stability and dissociative half-life of the ternary complex can directly correlate with the efficiency of intracellular target degradation, highlighting the critical importance of measuring kinetics in drug discovery [45].
The determination of association and dissociation rate constants via SPR is a cornerstone of modern molecular interaction analysis. While the 1:1 Langmuir model provides a fundamental framework, SPR is a versatile technique capable of probing complex interactions, including ternary complexes with significant therapeutic relevance. The kinetic parameters kₐ and kᵈ offer profound insights into the dynamics of molecular binding that equilibrium affinity constants alone cannot provide, enabling researchers to better understand biological mechanisms and engineer molecules with optimized binding profiles for therapeutic and diagnostic applications.
Surface Plasmon Resonance (SPR) has become a cornerstone technique in biophysical interaction analysis, providing a label-free method for studying molecular binding events in real-time. Within drug discovery and basic research, quantitatively determining the strength of an interaction between a ligand and an analyte is paramount. This is most frequently expressed as the equilibrium dissociation constant (KD), a crucial parameter that defines the concentration of analyte required to occupy half of the available binding sites on a ligand at equilibrium. A lower KD value indicates a higher binding affinity. SPR is uniquely capable of providing not only this steady-state affinity constant but also the individual kinetic rate constants—the association rate (ka) and dissociation rate (kd)—that define the dynamics of the interaction, from which KD can be derived as kd/ka [21] [46]. This guide details the core principles, methodologies, and data analysis techniques for robust KD determination using SPR, framed within the context of foundational SPR research.
Core Principles of SPR Technology
The SPR measurement principle is based on detecting changes in the refractive index near a sensor chip surface. In practice, one binding partner (the ligand) is immobilized on this surface, while the other (the analyte) is flowed over it in solution. As analyte molecules bind to the ligand, the mass at the surface increases, causing a proportional change in the refractive index that is detected as a shift in the SPR resonance angle (ϴ). This response is measured in resonance units (RU) and plotted in real-time to generate a sensorgram, a kinetic plot that tracks the entire binding event [21] [47].
The resulting sensorgram displays distinct phases:
- Association: When analyte is injected and binds to the ligand, causing an increase in RU.
- Equilibrium: The point where the binding and dissociation rates are equal, leading to a plateau in the sensorgram.
- Dissociation: When the analyte injection stops and buffer is flowed, causing a decrease in RU as the complex dissociates [21].
Experimental Design and Methodologies
A well-designed experiment is critical for generating reliable KD values. The following protocols outline the key steps.
Ligand Immobilization
The first critical step is attaching the ligand to the sensor chip surface. The chosen method can significantly impact the activity of the ligand and the quality of the data [21].
- Covalent Coupling (e.g., CMS Chip): A standard method using amine coupling, where the carboxymethylated dextran surface is activated with a mixture of N-hydroxysuccinimide (NHS) and N-ethyl-N'-(3-dimethylaminopropyl)carbodiimide (EDC). The ligand is then injected, coupling via primary amines, and remaining reactive groups are blocked with ethanolamine [21] [46]. The real-time process shows distinct phases of activation, coupling, and blocking [46].
- Capture Methods: These offer a more uniform orientation. Common approaches include:
- His-tag capture on an NTA chip.
- Biotin-streptavidin interaction.
- Antibody-based capture (e.g., using Protein A or Protein G chips) [21].
The immobilization level must be optimized; too high a density can cause mass transport effects, while too low a density may yield a weak signal [48].
Analyte Binding and Regeneration
Once the ligand is stably immobilized, the analyte is injected in a series of concentrations across the ligand surface and a reference surface.
- Running Buffer: The buffer must be optimized for the specific interaction, with a pH that maintains biological relevance. If analytes are dissolved in organic solvents like DMSO, the DMSO concentration must be matched in all solutions and the running buffer to prevent refractive index artifacts [21] [48].
- Regeneration: To reuse the ligand surface for multiple analyte injections, a regeneration solution is used to disrupt the binding interaction without denaturing the ligand. Finding a suitable regeneration condition—ranging from mild (e.g., 2 M NaCl) to harsh (e.g., 10 mM Glycine pH 2.0)—is often an empirical process [21]. It is essential to ensure the ligand is fully regenerated between cycles [48].
Data Analysis and KD Calculation
Analyzing the sensorgram data allows for the extraction of kinetic and steady-state constants. The following table summarizes the core parameters obtained from SPR analysis.
Table 1: Core Binding Parameters Measured by SPR
| Parameter | Symbol | Description | Significance |
|---|---|---|---|
| Association Rate Constant | ka (M⁻¹s⁻¹) | The rate at which the analyte binds to the ligand. | Defines how quickly a complex forms. |
| Dissociation Rate Constant | kd (s⁻¹) | The rate at which the analyte-ligand complex breaks apart. | Defines the stability or longevity of the complex. |
| Equilibrium Dissociation Constant | KD (M) | The analyte concentration at which half the ligand sites are occupied at equilibrium. KD = kd / ka. | The primary measure of binding affinity. |
| Maximum Binding Capacity | Rmax (RU) | The theoretical response at saturation, when all ligand sites are occupied. | Used for curve fitting and validating the binding model. |
Kinetic Analysis
This is the most powerful feature of SPR. The sensorgram data from a series of analyte concentrations is globally fitted to a binding model, typically a 1:1 Langmuir interaction, to simultaneously determine ka and kd. The KD is then calculated from the ratio of these rates [21] [46]. This method is highly accurate when the association and dissociation phases are well-defined and the data is of high quality.
Steady-State Analysis
When kinetics are too fast to measure accurately or a simpler analysis is sufficient, the steady-state or equilibrium response can be used. The average response at the equilibrium plateau for each analyte concentration is plotted against the concentration. This binding isotherm is then fitted to a non-linear regression model to derive the KD directly [21]. For this analysis, it is recommended to use 8-10 analyte concentrations to properly define the saturation curve [48].
Practical Application: A Case Study in Drug Discovery
To illustrate the practical application of SPR for KD determination, a recent study investigated the binding affinity of synthetic cannabinoids (SCs) to the CB1 receptor [46]. The experimental workflow and logical relationships for such a study can be summarized as follows:
In this study, the CB1 receptor was immobilized on a CM5 chip, and ten different SCs were flowed as analytes. The resulting KD values, determined through kinetic analysis, revealed clear structure-affinity relationships (SAR). For instance, indazole-based SCs consistently showed higher affinity (lower KD) than their indole-based counterparts [46]. The key quantitative findings are summarized below.
Table 2: Experiment Results - KD Values of Synthetic Cannabinoids to CB1 Receptor [46]
| Synthetic Cannabinoid | Parent Core | KD Value (M) | Relative Affinity |
|---|---|---|---|
| FUB-AKB-48 | Indazole | 1.571 × 10⁻⁶ | Highest |
| 5F-MDMB-PINACA | Indazole | 3.706 × 10⁻⁶ | Very High |
| MDMB-4en-PINACA | Indazole | 5.786 × 10⁻⁶ | Very High |
| AB-CHMINACA | Indazole | 6.458 × 10⁻⁶ | Very High |
| 5F-AKB-48 | Indazole | 8.287 × 10⁻⁶ | High |
| STS-135 | Indole | 1.770 × 10⁻⁵ | Medium |
| FDU-PB-22 | Indole | 1.844 × 10⁻⁵ | Medium |
| MAM-2201 | Indole | 2.293 × 10⁻⁵ | Medium |
| AMB-4en-PICA | Indole | 3.295 × 10⁻⁵ | Low |
| JWH-018 | Indole | 4.346 × 10⁻⁵ | Lowest |
The Scientist's Toolkit: Essential Research Reagents and Materials
Successful SPR experiments require careful selection of materials and reagents. The following table details key components used in the featured case study and their functions.
Table 3: Research Reagent Solutions for SPR Experiments
| Reagent / Material | Function in the Experiment |
|---|---|
| CM5 Sensor Chip | A carboxymethylated dextran matrix for covalent immobilization of ligands via amine coupling [21] [46]. |
| NHS/EDC Mixture | Activation chemistry for the CM5 chip surface; cross-links the ligand to the dextran matrix [21] [46]. |
| Ethanolamine HCl | A blocking agent used to deactivate and quench any remaining reactive ester groups on the chip surface after ligand immobilization [21] [46]. |
| HEPES Buffered Saline | A common running buffer (e.g., 10 mM HEPES pH 7.4, 150 mM NaCl) that maintains a stable pH and ionic strength during analyte injections [21]. |
| Nanodiscs | Lipid bilayer scaffolds used to present membrane-bound receptors (like CB1) in a more native-like environment for interaction studies [21]. |
| Regeneration Solutions | Buffers (e.g., 2 M NaCl, 10 mM Glycine pH 2.0) used to break the analyte-ligand complex without damaging the ligand, allowing for chip re-use [21]. |
Troubleshooting and Best Practices for Publication-Quality Data
To ensure data credibility and readiness for publication, researchers must be vigilant for common artefacts and adhere to best practices.
Evaluating Binding Curves: An ideal sensorgram should have association and dissociation phases that follow a single exponential, with curves that are well-spaced across different analyte concentrations. Common issues to identify and correct include:
- Mass Transport Effects: When the rate of analyte diffusing to the surface is slower than the binding rate, leading to a lack of curvature in the association phase. Remedied by reducing ligand density, increasing flow rate, or using higher analyte concentrations [48].
- Non-Specific Binding (NSB): When the analyte binds to the chip surface rather than the ligand. It is essential to run a control on a reference surface without ligand to test for and minimize NSB [48].
- Bulk Shift: A square-shaped signal caused by a difference in refractive index between the running buffer and the analyte buffer. Corrected by perfectly matching the buffer composition, including DMSO concentration [48].
Data Presentation for Publication: To enhance credibility, include the corrected raw data with the fitting model overlaid. Explicitly state the use of a reference surface, detail all experimental conditions (instrument, sensor chip, immobilization level, flow rates, buffers), and make the raw data available as supplemental information [48]. Experiments should be performed in duplicate or triplicate to demonstrate reproducibility and calculate standard deviations [48].
The determination of the equilibrium dissociation constant (KD) using Surface Plasmon Resonance is a powerful and versatile methodology that provides critical insights into molecular interactions. By following rigorous experimental design, from strategic ligand immobilization to meticulous data analysis, researchers can obtain reliable kinetic and affinity parameters. As demonstrated in the case of synthetic cannabinoid binding, SPR is capable of not only quantifying affinity but also elucidating detailed structure-activity relationships. Mastery of this technique, including adherence to best practices for troubleshooting and data presentation, makes it an indispensable tool in the fundamental research and drug development workflow.
Surface Plasmon Resonance (SPR) is a label-free optical biosensing technology that enables the real-time monitoring of biomolecular interactions [24] [49]. The fundamental principle relies on the excitation of surface plasmon polaritons—electromagnetic waves propagating at a metal-dielectric interface—typically using a prism-coupled Kretschmann configuration [49]. When light incident at a specific angle interacts with free electrons in a thin metal layer (usually gold), it generates an evanescent field that is exquisitely sensitive to refractive index changes within approximately 300 nanometers of the sensor surface [49]. This phenomenon makes SPR particularly powerful for quantifying binding events between biological molecules without requiring fluorescent or radioactive labels.
The versatility of SPR technology has established its critical importance in two distinct yet vital application domains: drug discovery and clinical diagnostics. In Fragment-Based Drug Discovery (FBDD), SPR provides the sensitivity needed to detect weak interactions between low molecular weight fragments and therapeutic targets [50] [51] [52]. Conversely, in pathogen detection, SPR platforms are engineered for maximum sensitivity to identify trace amounts of pathogen-specific biomarkers in clinical samples [53] [54]. This technical guide explores the specific methodologies, experimental protocols, and advanced applications of SPR in these two fields, emphasizing both their specialized requirements and shared technological foundations.
SPR in Fragment-Based Drug Discovery (FBDD)
The FBDD Workflow and SPR's Role
Fragment-Based Drug Discovery is a strategic approach that identifies small, low molecular weight compounds (<300 Da) as starting points for drug development [51] [52]. These fragments bind weakly to target proteins but exhibit high "ligand efficiency," serving as foundational scaffolds that can be optimized into potent drug candidates through iterative structure-guided chemistry [51]. SPR technology is integral to this process, primarily because of its ability to quantify these weak, transient interactions in real-time, which is challenging for other biophysical methods.
The following diagram illustrates the standard FBDD workflow with integrated SPR screening:
The FBDD process begins with a carefully designed fragment library, typically containing 500-3000 compounds with molecular weights below 300 Da [52]. SPR screening follows, where fragments are injected over immobilized target protein at high concentrations (100-500 μM) to detect weak binding (affinities typically in mM-μM range) [52]. Confirmed hits undergo rigorous validation using orthogonal biophysical methods such as X-ray crystallography or NMR to rule out false positives and determine binding modes [51]. The final stages involve fragment-to-lead optimization through structure-guided medicinal chemistry, enhancing affinity and drug-like properties while maintaining favorable ligand efficiency [51] [52].
Key Experimental Protocols for SPR in FBDD
Target Immobilization and Fragment Screening
Successful fragment screening requires careful experimental design to maximize sensitivity while minimizing non-specific binding. The following protocol outlines a standard approach for SPR-based fragment screening:
Sensor Surface Preparation:
- Surface Selection: Choose appropriate sensor chip chemistry based on target properties. CMS chips with carboxymethylated dextran are standard for amine coupling [49].
- Target Immobilization: Employ amine coupling chemistry for protein targets: (1) Activate carboxyl groups with EDC/NHS mixture (7 min, 25°C), (2) Dilute protein to 10-50 μg/mL in sodium acetate buffer (pH 4.0-5.5), (3) Inject protein until desired immobilization level is reached (5000-10,000 RU for fragment screening), (4) Block remaining activated groups with ethanolamine (7 min) [52].
- Reference Surface: Prepare a reference flow cell with activated and blocked surface without protein to correct for buffer effects and non-specific binding.
Fragment Screening Conditions:
- Running Buffer: Use HBS-EP (10 mM HEPES, 150 mM NaCl, 3 mM EDTA, 0.005% v/v Surfactant P20, pH 7.4) or phosphate buffer with 1-5% DMSO to maintain fragment solubility [52].
- Screening Parameters: Inject fragments as single concentrations (100-500 μM) for 30-60 seconds at 30 μL/min flow rate, followed by 60-120 seconds dissociation monitoring [52].
- Regeneration: When needed, use mild regeneration conditions (1-2 second pulse of 2-5 mM NaOH or HCl) to remove persistently bound fragments without damaging target activity [54].
Data Analysis and Hit Validation
Primary Screening Analysis:
- Response Criteria: Identify positive hits as fragments producing significant response (>50% theoretical Rmax) that exceeds 3× standard deviation of buffer blank injections [52].
- Binding Level: Expect low response units (5-50 RU) for fragment binding due to small molecular size.
Hit Confirmation:
- Dose-Response Analysis: For confirmed hits, perform steady-state affinity measurements using a concentration series (0.5-500 μM) to determine Kd values [52].
- Kinetic Characterization: When binding responses permit, extract association (kₒₙ) and dissociation (kₒff) rate constants using 1:1 binding model, recognizing that fragments often exhibit fast on/off rates [52].
- Selectivity Assessment: Implement counter-screens against mutant proteins or unrelated targets to establish binding specificity [50] [52].
FBDD Success Stories and Key Reagents
SPR-driven FBDD has generated numerous clinical successes, including eight FDA-approved drugs and over 50 clinical candidates [51] [52]. Notable examples include Vemurafenib (BRAF inhibitor for melanoma) and Venetoclax (BCL-2 inhibitor for leukemia) [51]. Recent advancements include the discovery of pan-RAS inhibitors through FBDD, addressing a historically challenging oncology target [50]. Additionally, covalent fragment approaches have emerged for targeting previously undruggable proteins, combining traditional FBDD with targeted covalent inhibition [50].
Table 1: Essential Research Reagent Solutions for SPR in FBDD
| Reagent/Solution | Function in FBDD | Application Notes |
|---|---|---|
| CMS Sensor Chips | Carboxymethylated dextran surface for protein immobilization | Standard for amine coupling; suitable for most soluble protein targets [49] |
| NTA Sensor Chips | Nickel chelation surface for His-tagged protein capture | Reversible capture; ideal for targets requiring frequent surface regeneration [49] |
| EDC/NHS Mixture | Crosslinking reagents for carboxyl group activation | Critical for amine coupling protocol; fresh preparation recommended [49] |
| Ethanolamine-HCl | Blocking solution for deactivating excess reactive groups | Standard quenching agent after immobilization [49] |
| HBS-EP Buffer | Standard running buffer with surfactant | Reduces non-specific binding; maintains stable baseline [52] |
| Reference Protein | Well-characterized binder for system suitability testing | Validates sensor surface activity and assay performance [52] |
SPR in Rapid Pathogen Detection:Mycobacterium tuberculosis
SPR Biosensor Design for Tuberculosis Diagnostics
The application of SPR biosensing for Mycobacterium tuberculosis (MTB) detection addresses critical limitations of conventional diagnostic methods, which often suffer from prolonged turnaround times (2-4 weeks for culture), low sensitivity, or requirement for sophisticated laboratory infrastructure [55] [54]. SPR platforms developed for TB detection leverage various sensing strategies, including nucleic acid hybridization for MTB complex-specific genes, antibody-antigen interactions for lipoarabinomannan detection, and whole-cell capture using mycobacteria-specific ligands [53] [54].
Recent advances in SPR biosensor design have demonstrated dramatically improved sensitivity through innovative material combinations. A 2025 study reported a multilayered SPR configuration (SiO₂/CaF₂/Ag/AlON/BP) that achieves exceptional angular sensitivity of 615.33 deg/RIU, enabling precise detection within the biological refractive index range of 1.29 to 1.35 [53]. This enhanced sensitivity is critical for detecting low abundance TB biomarkers in clinical samples, potentially enabling earlier diagnosis and intervention.
The following diagram illustrates a representative SPR biosensor configuration for MTB detection:
Experimental Protocol for SPR-based MTB Detection
DNA Hybridization-Based Detection
This protocol adapts the approach developed by Duman and Piskin (2010) for specific detection of MTB complex DNA, with enhancements from recent biosensor designs [53] [54]:
Sensor Surface Functionalization:
- Surface Cleaning: Treat gold sensor surfaces with UV/ozone cleaning for 15-20 minutes to remove organic contaminants [54].
- Probe Immobilization: Prepare thiol-modified oligonucleotide probes (20-25 mer) complementary to MTB complex-specific sequences (e.g., IS6110 insertion element). Dilute probes to 1 μM in immobilization buffer (1 M KH₂PO₄, pH 3.8). Inject over clean gold surface for 2-4 hours at 5 μL/min flow rate to form self-assembled monolayer [54].
- Surface Blocking: Treat with 1 mM 6-mercapto-1-hexanol (MCH) for 30 minutes to displace non-specifically adsorbed DNA and orient probes upright [54].
Sample Processing and Hybridization:
- DNA Extraction: Isolate DNA from sputum samples using standard commercial kits with mechanical lysis to break mycobacterial cell walls.
- Target Amplification: Apply asymmetric PCR or isothermal amplification to generate single-stranded target DNA without fluorescent labeling.
- Hybridization Conditions: Dilute amplified target in hybridization buffer (150 mM NaCl, 20 mM Na₂HPO₄, 0.1 mM EDTA, 0.005% Tween 20, pH 7.4). Inject over sensor surface for 5-10 minutes at 10-20 μL/min flow rate [54].
- Regeneration: Regenerate surface with 2.5 mM HCl pulse (30 seconds) for multiple assay cycles without significant signal degradation [54].
Data Analysis and Performance Validation
Signal Interpretation:
- Positive Identification: Significant shift in resonance angle/position (>3× baseline noise) relative to negative controls indicates positive hybridization.
- Quantification: Construct calibration curve using known concentrations of synthetic MTB DNA targets (0.001-1 μM) to estimate target concentration in clinical samples.
Analytical Performance:
- Sensitivity: The optimized protocol detects specific DNA hybridization at 0.05 μM concentration, with mass detection limit of 30 ng/μL [54].
- Specificity: Include control channels with non-complementary probes and specifically target M. gordonae sequences to discriminate from environmental mycobacteria [54].
- Stability: Properly prepared sensors maintain functionality for ≥12 weeks when stored in vacuum at room temperature in dark conditions [54].
Performance Comparison and Key Reagents
SPR biosensors for TB detection demonstrate competitive performance compared to conventional diagnostic methods, particularly in speed and potential for point-of-care application. The following table summarizes performance metrics for representative SPR platforms in TB detection:
Table 2: Performance Comparison of SPR Biosensors for MTB Detection
| Biosensor Configuration | Detection Target | Sensitivity | Assay Time | Key Advantage |
|---|---|---|---|---|
| Multilayer SPR [53] | Whole Cell (Refractive Index) | 615.33 deg/RIU angular sensitivity | Minutes | Maximum sensitivity; label-free |
| DNA Hybridization [54] | IS6110 DNA sequence | 0.05 μM (30 ng/μL) | <2 hours | High specificity for MTB complex |
| Portable SPR [54] | MTB complex vs. M. gordonae | 0.05 μM | <2 hours | Discrimination from NTM; portability |
| Conventional Culture [55] | Viable bacteria | 10-100 CFU/mL | 2-4 weeks | Gold standard; provides viability |
| Xpert MTB/RIF [55] | DNA & drug resistance | 95% LOD: 131 CFU/mL | <2 hours | WHO-endorsed; integrated PCR |
Table 3: Essential Research Reagent Solutions for SPR in MTB Detection
| Reagent/Solution | Function in MTB Detection | Application Notes |
|---|---|---|
| Thiolated DNA Probes | Target capture elements | Complementary to MTB-specific sequences (e.g., IS6110, 16S rRNA) [54] |
| 6-Mercapto-1-hexanol (MCH) | Surface blocking agent | Reduces non-specific adsorption; improves probe orientation [54] |
| Hybridization Buffer | Optimal binding conditions | Contains salts for ionic strength; surfactant to minimize aggregation [54] |
| Regeneration Solution | Surface renewal | Mild acid (2.5 mM HCl) effectively dissociates DNA duplexes [54] |
| DNA Extraction Kits | Target isolation from samples | Must include mechanical disruption for mycobacterial cell wall [55] |
| Amplification Reagents | Signal enhancement | Isothermal amplification suitable for resource-limited settings [55] |
Technological Convergence and Future Perspectives
The ongoing evolution of SPR technology demonstrates remarkable convergence between its applications in basic drug discovery and clinical diagnostics. Recent advances in high-throughput SPR instrumentation now enable parallel screening of fragments against multiple targets simultaneously, revealing selectivity patterns and promising starting points for chemical optimization [50]. These technological improvements benefit both FBDD and pathogen detection through increased throughput, reduced sample consumption, and enhanced data quality.
The integration of artificial intelligence and machine learning with SPR data analysis represents another significant frontier. In FBDD, AI-driven approaches are being applied to predict binding modes and optimize fragment elaboration [51]. Similarly, in diagnostics, pattern recognition algorithms can enhance the discrimination of specific binding signals from complex background matrices in clinical samples [56]. These computational advances complement hardware improvements to push the sensitivity and information content of SPR measurements.
Future developments will likely focus on increasing the accessibility of SPR technology through miniaturized, portable systems that maintain analytical performance while reducing cost and operational complexity. Such advancements promise to further expand SPR applications from central laboratories to point-of-care diagnostic settings and smaller research facilities, ultimately accelerating both drug discovery journeys and patient diagnosis timelines.
Solving Common SPR Challenges: A Troubleshooting and Optimization Guide
Surface Plasmon Resonance (SPR) is a powerful, label-free technology for the real-time monitoring of biomolecular interactions, playing a critical role in life sciences, pharmaceutics, and drug discovery [24]. The fundamental output of an SPR experiment is the sensorgram, a plot of response units (RU) against time, where a stable baseline is the essential foundation for obtaining accurate kinetic and affinity data. Baseline issues—drift, noise, and instability—represent some of the most frequent challenges in SPR experimentation, potentially compromising data quality and leading to erroneous conclusions. A thorough understanding of these phenomena is therefore a fundamental requirement for SPR research. This guide provides an in-depth technical examination of baseline anomalies, offering researchers systematic methodologies for their diagnosis and resolution.
Defining Baseline Anomalies: Characteristics and Causes
A pristine SPR baseline is characterized by a flat, stable signal with minimal high-frequency fluctuations when only the running buffer is passing over the sensor chip. Deviations from this ideal state manifest as specific anomalies, each with distinct visual characteristics and underlying causes.
Baseline Drift
Drift is characterized by a gradual, monotonic increase or decrease in the baseline signal over time [57]. It is typically a sign of a system or surface that is not fully equilibrated.
- Primary Causes:
- Sensor Chip Equilibration: Newly docked sensor chips or surfaces freshly after ligand immobilization require time to rehydrate and equilibrate with the running buffer. Chemicals from immobilization procedures wash out slowly, causing drift [57].
- Buffer Incompatibility: Changing the running buffer without sufficient system priming causes mixing of buffers with different refractive indices, resulting in a drifting baseline as the system reaches a new equilibrium [57].
- Start-up Effects: Initiating fluid flow after a period of stagnation can induce drift as the sensor surface acclimates to the new flow conditions. This typically levels out within 5–30 minutes [57].
Baseline Noise
Noise appears as high-frequency, low-amplitude fluctuations superimposed on the baseline signal. While some noise is inherent to the instrument, excessive noise obscures small binding signals and reduces data quality.
- Primary Causes:
- Air Bubbles: Small air bubbles in the fluidic system, often from inadequately degassed buffers stored at 4°C, cause sharp spikes and increased noise [57].
- Pressure Fluctuations: The SPR system is highly sensitive to pressure differences within the fluidic path, which cause abrupt response changes [57].
- Particulate Contamination: Impurities in the buffer or samples can introduce transient noise as they pass through the flow cell.
- Electronic Noise: This originates from the instrument's optical detection system or associated electronics.
Baseline Instability (Jumps and Spikes)
Instability refers to sudden, large, and often abrupt shifts in the baseline signal. Unlike drift, these are not gradual and can occur at specific points in an experiment.
- Primary Causes:
- Air Spikes: The most common cause, resulting from small air bubbles entering the flow cell [57].
- Pump Events: The pump refilling (e.g., during an injection) can stop the flow for seconds, creating a noticeable spike in the sensorgram [57].
- Improper Regeneration: Harsh regeneration conditions can partially damage the ligand or the sensor surface itself, leading to a permanent drop in the baseline response and often introducing drift in subsequent cycles [42] [58].
Table 1: Summary of Common Baseline Anomalies and Their Origins
| Anomaly Type | Visual Characteristic | Common Causes |
|---|---|---|
| Drift | Gradual, sustained increase or decrease in signal | Incomplete surface equilibration; Buffer change; Start-up after flow stall [57] |
| Noise | High-frequency, low-amplitude fluctuations | Air bubbles; Pressure fluctuations; Particulate contamination [57] |
| Spikes/Jumps | Abrupt, large shifts in signal | Air bubbles; Pump refill events; Mechanical disturbances [57] |
A Systematic Workflow for Diagnosing Baseline Issues
Effective troubleshooting requires a logical, step-by-step approach to isolate the root cause of baseline problems. The following diagnostic workflow provides a structured methodology for researchers.
Diagram 1: Systematic diagnostic workflow for SPR baseline issues.
Foundational Checks: Buffer and Fluidics
The vast majority of baseline issues originate from the solutions and fluidic path. These should always be the first point of investigation.
Buffer Preparation and Handling:
- Prepare Fresh Daily: Buffers should be prepared fresh each day to prevent microbial growth or chemical degradation [57].
- Filtration and Degassing: Always filter buffers through a 0.22 µm filter and degas thoroughly before use. Buffers stored at 4°C contain more dissolved gas and are prone to forming bubbles; warm to room temperature and degas immediately before use [57].
- Hygiene: Do not add fresh buffer to old stock. Use clean, sterile bottles for storage. Transfer an aliquot to a new bottle for degassing and immediate use [57].
- Detergent Addition: Add detergents like Tween-20 after filtering and degassing to prevent foam formation [57].
Fluidic System Inspection:
- Prime Thoroughly: After any buffer change, prime the system extensively to eliminate the previous buffer completely. Failure to do so results in mixing and a wavy baseline due to refractive index differences [57].
- Check for Bubbles: Visually inspect the buffer lines, degassing unit, and injection system for air bubbles.
- Ensure Tight Connections: Verify that all fluidic connections are secure to prevent leaks that can cause pressure fluctuations and air ingress.
Sensor Surface and Instrument Evaluation
If buffer and fluidics are confirmed to be optimal, the investigation should focus on the sensor surface and the instrument itself.
Sensor Surface Equilibration:
- Allow Sufficient Time: A new sensor chip or a freshly immobilized surface often requires an extended period (sometimes overnight) of buffer flow to fully equilibrate and stabilize [57].
- Use Start-up Cycles: Incorporate at least three start-up cycles at the beginning of an experimental method. These cycles should be identical to sample cycles but inject running buffer instead of analyte. This "primes" the surface and stabilizes it before data collection; these cycles should not be used for data analysis [57].
Instrument Maintenance and Calibration:
- System Cleaning: Follow the manufacturer's recommended procedures for cleaning the Integrated Fluidic Cartridge (IFC) and optics. Contamination here can cause high noise and drift.
- Calibration: Perform routine instrument calibration as specified in the user manual. A misaligned optical system or uncalibrated detector can lead to instability and poor performance [57].
Experimental Protocols for Mitigation and Optimization
Once the root cause is identified, specific protocols can be implemented to resolve the issue and prevent its recurrence.
Protocol for Minimizing Baseline Drift
Objective: To achieve a stable baseline with a drift rate of < 1 RU/minute.
- Pre-equilibration: After immobilization, dock the sensor chip and initiate a continuous flow of running buffer at the experimental flow rate. Monitor the baseline for 30-60 minutes. For surfaces with severe drift, this may need to be extended to several hours or overnight [57].
- Start-up Cycles: Program your experimental method to include a minimum of three start-up cycles. These cycles should include a buffer injection, dissociation phase, and a regeneration injection (if used). This conditions the surface before analytical cycles [57].
- Buffer Matching: Ensure the running buffer and sample buffer (including additives like DMSO) are identical. Mismatched buffers cause bulk refractive index shifts, which can manifest as drift after injection [42].
- Double Referencing: This data processing technique is critical for compensating for residual drift and bulk effects.
- Step 1: Reference Surface Subtraction. Subtract the signal from a reference flow cell (with no ligand or an irrelevant ligand) from the active flow cell signal. This removes most of the bulk effect and system-related drift [57].
- Step 2: Blank Subtraction. Subtract the averaged response from multiple blank injections (running buffer) from the analyte injection signals. This corrects for differences between the reference and active surfaces and further refines the baseline [57]. Blank cycles should be spaced evenly throughout the experiment, approximately one every five to six analyte cycles [57].
Protocol for Reducing Noise and Spikes
Objective: To achieve a system noise level of < 1 RU, as demonstrated in a properly functioning system [57].
- Rigorous Buffer Prep:
- Filter 1-2 liters of buffer through a 0.22 µm membrane into a clean, sterile bottle [57].
- Degas for 20-30 minutes with continuous stirring.
- Add non-ionic surfactant (e.g., 0.05% Tween 20) after degassing.
- System Purge:
- Prime the system at least three times with the freshly prepared, degassed buffer.
- After priming, flow the buffer for 15-30 minutes to allow the system to stabilize before starting the experiment.
- Baseline Noise Test:
- Once the baseline is stable, perform several consecutive buffer injections.
- Observe the sensorgram. The average baseline response should be stable, and the noise level during the injection plateau should be very low (< 1 RU) [57]. This serves as a quality control for the entire system.
Table 2: Troubleshooting Guide for Persistent Baseline Issues
| Symptom | Possible Cause | Recommended Solution |
|---|---|---|
| Persistent high drift after immobilization | Sensor surface not equilibrated; Unstable ligand immobilization | Extend pre-equilibration time with buffer flow; Check immobilization chemistry and ligand stability [57]. |
| Sudden baseline jumps followed by drift | Harsh regeneration damaging the surface | Optimize regeneration conditions; Start with mild solutions and increase stringency gradually; Use short contact times (100-150 µL/min) [42]. |
| High noise across all flow cells | Contaminated buffer or fluidics; Air in the system; Instrument issue | Re-prepare fresh, filtered, degassed buffer; Purge fluidic lines; Perform instrument maintenance and calibration [57]. |
| Noise and drift in one specific flow cell | Defective sensor chip or localized contamination | Replace the sensor chip; Inspect the specific flow cell for visible defects [57]. |
The Scientist's Toolkit: Essential Reagents and Materials
The following table details key reagents and materials essential for achieving and maintaining a stable SPR baseline.
Table 3: Essential Research Reagent Solutions for Stable SPR Baselines
| Item | Function & Importance | Technical Notes |
|---|---|---|
| High-Purity Water | Solvent for all buffers; impurities cause non-specific binding and noise. | Use ultrapure water (18.2 MΩ·cm resistivity) from a reliable purification system. |
| Analytical Grade Buffers | Provides stable pH and ionic strength; crucial for interaction integrity. | Common buffers: HEPES, PBS. Prepare fresh daily and filter. |
| Non-ionic Surfactant (e.g., Tween-20) | Reduces non-specific binding (NSB) by disrupting hydrophobic interactions. | Typically used at 0.005%-0.05%. Add after filtering and degassing to prevent foam [42] [58]. |
| Blocking Agents (e.g., BSA, Ethanolamine) | Blocks remaining reactive groups on the sensor surface after immobilization. | Minimizes NSB. Ethanolamine is used after amine coupling; BSA can be added to analyte buffer [42] [58]. |
| Regeneration Solutions | Removes bound analyte without damaging the ligand. Critical for reusability. | Common solutions: Glycine-HCl (low pH), NaOH. Must be optimized for each interaction [42]. |
| Sensor Chips | The platform for ligand immobilization. Choice of chemistry is critical. | Select chip type (e.g., CM5 for carboxyl, NTA for His-tag) to match ligand and minimize NSB [42] [58]. |
Advanced Data Processing Techniques
For certain persistent issues, advanced data processing algorithms can help extract reliable data from suboptimal sensorgrams.
- Dynamic Baseline Adjustment: In fiber SPR sensing, where determining the resonance point can be challenging, algorithms like Particle Swarm Optimization (PSO) can be employed. PSO intelligently tracks the optimal dynamic baseline by finding the best combination of parameters (spectral width, measurement points), improving tracking ability and anti-noise performance, especially under conditions of light source fluctuation [59].
- Karhunen-Loeve (KL) Conversion: For specialized applications like Electrochemical SPR (EC-SPR), where the SPR curve becomes distorted due to light absorption, traditional minimum-point detection fails. KL conversion, similar to Principal Component Analysis, processes the entire SPR curve as a dataset to extract feature displacements dispersed across multiple angles, rather than relying on a single SPR angle. This method is particularly effective for non-uniform sensor surfaces [60].
A stable baseline is not merely a convenience in SPR research; it is a fundamental prerequisite for generating high-quality, publishable data on biomolecular interactions. As SPR technology continues to evolve and find new applications in drug discovery and biosensing [24], the principles of rigorous buffer management, meticulous experimental design, and systematic troubleshooting remain constant. By adhering to the protocols and diagnostic frameworks outlined in this guide, researchers can proactively address the challenges of baseline drift, noise, and instability, thereby ensuring the integrity and reliability of their SPR data.
Surface Plasmon Resonance (SPR) is a powerful, label-free analytical technique that enables the real-time monitoring of biomolecular interactions, making it indispensable in life sciences, pharmaceutics, and diagnostic development [24]. A key strength of SPR is its ability to determine both steady-state and kinetic binding affinities between interacting partners, from small molecules to entire cells [21] [61]. However, obtaining high-quality, interpretable data can be challenged by several common signal-related issues: a complete absence of signal (No Signal), a response that is too low for accurate quantification (Weak Signal), or a response that exceeds the instrument's detection range (Signal Saturation). This guide provides a structured, experimental approach to diagnosing and resolving these fundamental problems within the context of rigorous SPR research.
Core Principles of SPR Signal Generation
The SPR signal is an optical measurement reported in Resonance Units (RU), which is directly proportional to the mass concentration of analyte bound to the ligand immobilized on the sensor chip [61]. The signal arises from changes in the refractive index at the surface of the gold chip when binding events occur [21]. A typical sensorgram plots RU against time and displays distinct phases: a baseline (buffer flow), an association phase (analyte injection and binding), and a dissociation phase (buffer flow and complex breakdown) [21]. Understanding this expected profile is the first step in identifying anomalies.
The following workflow outlines a systematic approach to diagnosing and resolving the most common SPR signal issues:
Troubleshooting No Signal
A complete absence of signal indicates a fundamental failure in the binding event or its detection.
Diagnostic Procedures and Experimental Protocols
- Confirm System Operation: Verify that the analyte was injected correctly and that the microfluidic system is free of air bubbles or blockages [61]. Run a system performance test with a known standard interaction.
- Verify Ligand Immobilization: Check the immobilization level report. A successful immobilization should show a significant increase in RU from the baseline. If the immobilization level is zero or extremely low, the coupling chemistry may have failed [21].
- Assess Ligand Activity and Orientation: Even with successful immobilization, the ligand may be denatured or immobilized in a conformation that buries the binding site. Use a different immobilization strategy (e.g., amine coupling vs. tag capture) to orient the ligand favorably [21] [61]. For capture methods, ensure the tag is accessible and the ligand is native-folded.
- Validate Buffer Conditions: Ensure the running buffer is compatible with the interaction. The pH, ionic strength, and presence of essential co-factors (e.g., ions, ATP, Mg²⁺) can be critical for maintaining protein structure and binding capability [21]. Mismatched solvent conditions (e.g., DMSO percentage) between the running buffer and analyte sample can also cause significant refractive index disturbances and mask binding [21].
Resolving Weak Signal
A weak signal, characterized by a low response that is difficult to distinguish from noise, often stems from suboptimal mass transfer or insufficient binding sites.
Key Calculations and Optimization Strategies
The maximum expected response (Rmax) can be calculated using the formula [21]: Responsemax = (ResponseLigand × MassAnalyte) / MassLigand This calculation is crucial for experimental design. A low Rmax predicts a weak signal.
- Increase Ligand Density: If the calculated Rmax is low, immobilize more ligand to the sensor surface to increase the number of available binding sites [21] [61].
- Optimize Analyte Concentration: Inject a higher concentration of analyte. A series of concentrations should yield a concentration-dependent increase in response.
- Address Mass Transport Limitations: For large analytes like nanoparticles, which have slow diffusion coefficients, binding kinetics can be limited by the rate of analyte delivery to the chip surface. This can manifest as a weak and poorly fitting kinetic signal. To mitigate this, use higher flow rates and lower ligand densities [61].
- Chip Selection for Small Molecules: For analytes with a low molecular weight (<200 Da), the mass change upon binding is small. Specialized sensor chips with a high capacity for ligand immobilization (e.g., CM7) are designed to amplify the response for such applications [21].
Table 1: Strategies to Resolve Weak Signal Based on Analyte Type
| Analyte Type | Primary Cause of Weak Signal | Recommended Solution | Alternative Approach |
|---|---|---|---|
| Small Molecule | Low mass contribution per binding event | Use high-capacity chip (e.g., CM7) | Immobilize a fragment of the target to increase Rmax [21] |
| Protein | Low ligand density or inactive ligand | Optimize immobilization chemistry and density | Use oriented capture (e.g., His-tag, Biotin) to improve activity [21] |
| Nanoparticle (NanoRx) | Steric hindrance or mass transport limitation | Use a flat chip surface (e.g., C1 chip) [61] | Increase flow rate and reduce ligand density [61] |
Managing Signal Saturation
Signal saturation occurs when the binding response exceeds the instrument's linear detection range, often resulting in a flattened sensorgram at the top of the association phase, which precludes accurate kinetic analysis.
Experimental Corrections and Considerations
- Reduce Ligand Density: The most effective way to prevent saturation is to immobilize less ligand. This reduces the total number of binding sites and lowers the maximum achievable response (Rmax) [61].
- Dilute Analyte Concentration: Inject a lower concentration of analyte. This slows down the association rate and prevents the rapid saturation of all available binding sites.
- Shorten Association Time: If using a high concentration of analyte is necessary, reduce the contact time during injection to prevent the system from reaching full saturation.
- Confirm Specific Binding: Ensure that the massive signal is due to specific binding to the ligand and not non-specific adsorption to the sensor chip or the reference surface [61]. A properly prepared reference flow cell is essential for correcting this.
Table 2: Summary of Signal Problems, Causes, and Solutions
| Problem | Common Causes | Diagnostic Checks | Corrective Actions |
|---|---|---|---|
| No Signal | Ligand not immobilized;Inactive analyte;Critical buffer mismatch | Check immobilization level;Verify analyte activity with other methods;Check for DMSO/solvent mismatch [21] | Repeat coupling;Use a tagged ligand for oriented capture [21];Match buffer conditions exactly |
| Weak Signal | Low ligand density;Low analyte concentration;Mass transport limitation (large analytes) | Calculate expected Rmax [21];Inject a high analyte conc. as test;Check flow rate | Increase ligand immobilization;Use higher analyte conc.;Increase flow rate, use flatter chip [61] |
| Signal Saturation | Ligand density too high;Analyte concentration too high;Non-specific binding | Inspect sensorgram for flattened top;Check response on reference surface | Reduce ligand density;Use lower analyte conc.;Shorten injection time |
The Scientist's Toolkit: Essential Research Reagents and Materials
The following reagents and materials are critical for preparing and executing a robust SPR experiment aimed at avoiding common signal issues [21].
Table 3: Key Research Reagent Solutions for SPR Experiments
| Reagent/Material | Function in SPR Experiment | Application Notes |
|---|---|---|
| CM5 Sensor Chip | Gold chip with carboxymethylated dextran matrix; the most common chip for covalent immobilization of ligands via amine coupling [21] [61]. | Suitable for most protein/protein interactions. May cause steric hindrance for very large analytes [61]. |
| C1 Sensor Chip | Gold chip with a flat, 2-D-like surface and no dextran layer [61]. | Ideal for large analytes like nanoparticles or cells to prevent steric hindrance and mass transport issues [61]. |
| NTA Sensor Chip | Chip pre-immobilized with nitrilotriacetic acid (NTA) for capturing His-tagged ligands [21]. | Provides a uniform, oriented capture of ligands, helping to preserve activity. Requires Ni²⁺ or Co²⁺ charging. |
| EDC/NHS Chemistry | Cross-linking reagents (1-Ethyl-3-(3-dimethylaminopropyl)carbodiimide and N-Hydroxysuccinimide) used to activate carboxyl groups on the chip surface for covalent ligand attachment [21] [61]. | Standard for amine coupling. Freshly prepared solutions are critical for efficient activation. |
| HEPES Buffered Saline | A common running buffer (e.g., 10 mM HEPES pH 7.4, 150 mM NaCl) [21]. | Provides a physiologically relevant pH and ionic strength. Good buffering capacity without interfering with common interactions. |
| Regeneration Solution | A solution (e.g., 2 M NaCl, 10 mM Glycine pH 2.0-3.0) used to remove bound analyte from the immobilized ligand without damaging it [21]. | Allows for re-use of the sensor chip. Must be optimized for each specific ligand-analyte pair to be effective yet gentle. |
| Membrane Scaffold Protein (MSP) | Used to create lipid nanodiscs that can be immobilized on the chip to study membrane-protein or lipid interactions in a more native environment [21]. | Essential for incorporating specific lipids (e.g., Phosphatidic Acid) into a stable, soluble bilayer disc for SPR analysis [21]. |
Advanced Considerations: Mass Transport and Kinetic Analysis
For accurate kinetic analysis, it is vital to ensure that the observed binding rate is not limited by the diffusion of the analyte to the sensor surface. When mass transport is the rate-limiting step, the calculated association rate (kₐ) will be artificially low and the dissociation rate (k_d) may be inaccurate [61] [62].
Advanced mathematical modeling, such as using the Generalized Integral Transform Technique (GITT) to solve convective-diffusive-reaction equations coupled with the Markov Chain Monte Carlo (MCMC) method for parameter estimation, can be employed to deconvolute mass transport effects from the true binding kinetics [62]. Experimentally, using a lower ligand density and a higher flow rate can help minimize these limitations, ensuring that the measured rates reflect the actual biomolecular interaction [61].
In Surface Plasmon Resonance (SPR) research, the accurate measurement of biomolecular interactions is paramount. SPR is an optical technique that measures molecular interactions in real time by detecting changes in the refractive index on a sensor chip surface when a binding event occurs [4] [1]. The core challenge in obtaining high-quality binding data lies in distinguishing specific interactions between the ligand (immobilized molecule) and analyte (solubilized molecule) from non-specific binding (NSB) [63].
NSB refers to the adsorption of the analyte to the sensor surface through interactions other than the specific biological interaction of interest. This can include hydrophobic interactions, hydrogen bonding, Van der Waals forces, and electrostatic interactions [63]. When NSB occurs, it inflates the response units (RU) measured by the SPR instrument, leading to erroneous calculations of kinetic constants (association rate ka, dissociation rate kd) and equilibrium affinity (KD) [63]. Effectively combating NSB is therefore not merely an optimization step but a fundamental requirement for generating publication-quality data in drug development and basic research.
Mechanisms and Strategies for Reducing NSB
Non-specific binding fundamentally arises from physicochemical interactions between the analyte and the sensor surface. The primary mechanisms include:
- Hydrophobic Interactions: Occur between non-polar regions of biomolecules and hydrophobic surfaces on the sensor chip.
- Electrostatic Interactions: Attraction between positively charged residues on a protein and a negatively charged sensor surface (e.g., a carboxylated dextran matrix).
- Hydrogen Bonding: Polar groups on the analyte form hydrogen bonds with chemical groups on the sensor surface.
- Van der Waals Forces: Weak, non-covalent forces that can contribute to overall NSB [63] [64].
A preliminary test to determine the level of NSB involves running the analyte over a bare sensor surface without any immobilized ligand. A significant response indicates the presence of NSB that must be addressed before proceeding with specific binding experiments [63].
Buffer Additive Strategies
The most common and effective approach to minimize NSB involves optimizing the composition of the running buffer and sample diluent. The table below summarizes the primary buffer additive strategies.
Table 1: Buffer Additive Strategies to Combat Non-Specific Binding
| Strategy | Recommended Additive | Typical Working Concentration | Primary Mechanism of Action | Considerations |
|---|---|---|---|---|
| Protein Blockers | Bovine Serum Albumin (BSA) | 0.1-1% (w/v) | Shields the analyte from NSB by saturating non-specific sites on the surface and tubing. [63] | A globular protein with domains of varying charge densities. |
| Non-ionic Surfactants | Tween 20, Polysorbate 20 | 0.005-0.05% (v/v) | Disrupts hydrophobic interactions between the analyte and sensor surface. [4] [63] | A mild detergent; also prevents analyte loss to tubing and containers. |
| Salt Shielding | Sodium Chloride (NaCl) | 150-500 mM | Shields charged groups, reducing electrostatic-based NSB. [63] | High concentrations may salt out proteins or affect specific binding. |
| Polymeric Blockers | Polyethylene Glycol (PEG), Dextran | 0.1-1% (w/v) | Forms a hydrated, steric barrier that prevents non-specific adsorption. [65] | Molecular weight can influence effectiveness. |
The selection of the optimal additive depends on the physicochemical properties of the ligand and analyte, particularly their isoelectric point (pI), overall charge, and hydrophobicity. For instance, if the analyte is positively charged at a given pH, it may non-specifically bind to a negatively charged sensor surface; this can be mitigated by adjusting the buffer pH or increasing the ionic strength to shield the charges [63].
Table 2: Experimental Protocol for Buffer Additive Screening
| Step | Action | Purpose | Key Parameters |
|---|---|---|---|
| 1 | Prepare running buffers with different additives. | To empirically determine the most effective NSB reduction strategy. | Test one additive per buffer (e.g., Buffer A: 0.05% Tween 20, Buffer B: 1% BSA, Buffer C: 250 mM NaCl). |
| 2 | Immobilize ligand on one flow cell; leave another as a reference. | To create a specific binding surface and a control for NSB. | Follow standard amine coupling or capture coupling protocols. [4] |
| 3 | Inject analyte dissolved in the various running buffers. | To measure the level of specific binding and NSB under different conditions. | Use the same analyte concentration and injection parameters for all tests. |
| 4 | Analyze the response on the reference flow cell. | To directly quantify NSB for each buffer condition. | A lower reference cell signal indicates less NSB. |
| 5 | Compare the signal-to-noise ratio. | To select the optimal buffer condition. | The condition with the highest specific binding (sample cell) and lowest NSB (reference cell) is optimal. [63] |
Surface Preconditioning and Ligand Immobilization Optimization
Preconcentration: Enhancing Immobilization Efficiency
Preconcentration is a critical step prior to covalent immobilization (e.g., using Carboxyl sensors) that significantly increases the local density of the ligand on the sensor surface. This is achieved by adjusting the pH of the immobilization buffer to create opposite net charges on the sensor surface and the protein ligand, leading to electrostatic attraction and accumulation of the ligand near the surface [66].
This technique not only improves the efficiency of the subsequent covalent coupling but also allows researchers to use minimal amounts of precious protein samples to achieve a high immobilization level. A local ligand concentration of over 100 mg/mL can be achieved starting with a bulk solution of just 10 µg/mL [66].
Experimental Protocol: Preconcentration pH Scouting
The following detailed protocol allows for the determination of the optimal pH for preconcentration using a single sensor chip.
Table 3: Protocol for Preconcentration pH Scouting
| Step | Action | Details and Reagents |
|---|---|---|
| 1. Preparation | Dissolve ligand in acetate buffers of varying pH. | Prepare ligand at a low concentration (5-25 µg/mL) in 10 mM acetate buffers (e.g., pH 4.0, 4.5, 5.0, 5.5). Ensure identical ligand concentration in each buffer. [66] |
| 2. System Setup | Load a standard Carboxyl Sensor and condition. | Use a non-activated chip. Prepare 10 mM HCl as a regeneration solution. |
| 3. Ligand Injection | Inject the ligand solution at the highest pH. | Flow rate: 10 µL/min. Monitor the response; a large signal increase indicates strong electrostatic preconcentration. |
| 4. Surface Regeneration | Inject regeneration solution. | 10 mM HCl at 100 µL/min to strip the non-covalently bound ligand from the surface. |
| 5. Repeat | Repeat Steps 3 & 4 for each pH condition. | Test each buffer pH in duplicate for reliability. |
| 6. Analysis | Plot the response curves. | The optimal immobilization buffer is the one with the highest pH that still produces a large pre-concentration signal. [66] |
The Scientist's Toolkit: Essential Reagents for SPR Experiments
Table 4: Key Research Reagent Solutions for SPR
| Reagent / Supply | Function / Purpose | Examples / Notes |
|---|---|---|
| Sensor Chips | Provides the solid support for ligand immobilization. | Cytiva CM5 (carboxylated dextran); various surfaces (e.g., NTA for His-tag capture, Streptavidin for biotinylated ligands). [4] |
| Running Buffer | The liquid phase for transporting analyte over the ligand. | HBS-EP (10 mM HEPES, 150 mM NaCl, 3 mM EDTA, 0.05% surfactant P20) is a common starting point. [4] |
| Blocking Agents | Reduce NSB by saturating non-specific sites. | BSA, non-fat dry milk, casein, or proprietary commercial blockers. Avoid milk in biotin-streptavidin systems. [63] [67] |
| Surfactants | Reduce hydrophobic interactions and prevent analyte loss. | Tween 20 (Polysorbate 20) at 0.05% is standard. [4] [63] |
| Immobilization Buffers | Control pH and ionic strength for covalent coupling. | Acetate buffers (10-100 mM) for amine coupling; pH is selected based on ligand pI. [66] [65] |
| Regeneration Solutions | Dissociate bound analyte without damaging the ligand. | Low pH (e.g., 10 mM Glycine-HCl, pH 1.5-2.5), high salt, or specific chelators. Must be determined empirically. [65] |
Workflow and Data Interpretation
The following diagrams outline the logical workflow for addressing NSB and the key phases of an SPR sensorgram.
Diagram 1: NSB Troubleshooting Workflow. This flowchart outlines a systematic approach to diagnosing and mitigating non-specific binding in SPR experiments.
Diagram 2: Key Phases of an SPR Sensorgram. A typical sensorgram shows the response (RU) over time, corresponding to distinct phases of the binding interaction, which can be analyzed to extract kinetic and affinity constants [1].
Combating non-specific binding is a non-negotiable foundation for robust SPR research. By understanding its mechanisms and systematically applying strategies such as buffer optimization with additives like BSA and Tween 20, salt shielding, and careful surface preconditioning via preconcentration, researchers can significantly enhance data quality. The experimental protocols and troubleshooting workflows provided here offer a concrete path for scientists and drug development professionals to validate their molecular interaction data, ensuring that measured binding signals truly reflect the specific biology under investigation.
In Surface Plasmon Resonance (SPR) research, regeneration is not merely a procedural step but a critical determinant for experimental success and cost-effectiveness. SPR is an optical technique that measures molecular interactions in real time by detecting changes in the refractive index on a sensor chip surface when biomolecules bind [4]. The regeneration process involves removing bound analyte from the immobilized ligand after a binding cycle without permanently damaging the ligand activity, thereby allowing the same sensor surface to be reused for multiple experimental cycles [68] [69].
The strategic importance of robust regeneration protocols extends across the pharmaceutical and biotechnology sectors, where SPR is indispensable for characterizing binding kinetics, affinity, and specificity in drug discovery and biomolecular interaction analysis. Effective regeneration protocols significantly reduce consumable costs by extending sensor chip lifespan, enhance data quality by maintaining consistent surface binding properties across multiple cycles, and improve experimental throughput by enabling rapid screening of multiple analytes against the same ligand surface [70]. For researchers developing SPR methodologies within a broader thesis framework, mastering regeneration techniques represents a fundamental competency that bridges theoretical principles with practical experimental optimization.
Principles of Effective Regeneration
The Fundamental Challenge: Balancing Completeness and Ligand Integrity
The core challenge in regeneration protocol development lies in striking a precise balance between two competing objectives: complete analyte removal and preservation of ligand functionality. Regeneration buffers must be sufficiently harsh to disrupt all binding interactions between the ligand and analyte, yet sufficiently mild to maintain the ligand's structural integrity and binding capacity for subsequent experimental cycles [69]. This balance is complicated by the diverse nature of molecular interactions in biological systems, which may involve multiple binding forces requiring different disruption strategies.
Molecular interactions in SPR experiments are stabilized by various forces including electrostatic interactions, hydrophobic effects, hydrogen bonding, and van der Waals forces. Successful regeneration strategies target these specific interaction forces with appropriate chemical treatments [68]. The duration of regeneration buffer contact with the sensor surface represents another critical parameter, as prolonged exposure to harsh conditions may progressively degrade ligand activity even when short exposures are effective [70].
Assessing Regeneration Efficacy and Surface Stability
Evaluating regeneration success requires monitoring multiple parameters across several binding cycles. The primary indicator of effective regeneration is a stable baseline that returns to its original level after each regeneration cycle, demonstrating complete analyte removal without ligand damage [69]. The binding response upon subsequent analyte injections should remain consistent when the same analyte concentration is applied; significant deviation indicates either incomplete regeneration (response increases) or ligand degradation (response decreases) [69].
- Incomplete Regeneration: Results in residual analyte accumulation, manifested as a progressively rising baseline and increased binding responses with repeated cycles.
- Excessive Regeneration: Causes ligand denaturation or removal, evidenced by a declining baseline and reduced binding capacity over time.
Long-term surface stability must be assessed through multiple regeneration cycles (typically 10 or more) to establish protocol robustness, particularly for high-throughput applications requiring extensive surface reuse [70].
Regeneration Methodologies and Experimental Approaches
Systematic Development of Regeneration Protocols
The Cocktail Approach for Method Development
Andersson et al. developed a systematic "cocktail approach" to efficiently identify optimal regeneration conditions by targeting multiple binding forces simultaneously [68]. This methodology begins with preparing six stock regeneration solutions, each targeting different types of molecular interactions:
Table 1: Stock Solutions for Cocktail Regeneration Approach
| Solution Type | Composition | Target Interactions |
|---|---|---|
| Acidic | Equal volumes of oxalic acid, H₃PO₄, formic acid, and malonic acid (each 0.15 M), adjusted to pH 5.0 with NaOH | Electrostatic, hydrogen bonding |
| Basic | Equal volumes of ethanolamine, Na₃PO₄, piperazin, and glycine (each 0.20 M), adjusted to pH 9.0 with HCl | Electrostatic, hydrogen bonding |
| Ionic | KSCN (0.46 M), MgCl₂ (1.83 M), urea (0.92 M), guanidine-HCl (1.83 M) | Ionic, hydrophobic |
| Non-polar Solvents | Equal volumes of DMSO, formamide, ethanol, acetonitrile, and 1-butanol | Hydrophobic |
| Detergents | 0.3% (w/w) CHAPS, 0.3% (w/w) Zwittergent 3-12, 0.3% (v/v) Tween 80, 0.3% (v/v) Tween 20, 0.3% (v/v) Triton X-100 | Hydrophobic |
| Chelating | 20 mM EDTA | Metal-dependent interactions |
The experimental workflow involves mixing these stock solutions in various combinations, testing regeneration efficacy after analyte binding, and iteratively refining the composition based on performance.
Figure 1: Systematic Workflow for Developing Regeneration Protocols
Regeneration Solutions for Specific Interaction Types
Different molecular interactions require tailored regeneration approaches. The following table summarizes recommended regeneration conditions based on interaction characteristics:
Table 2: Regeneration Solutions for Specific Molecular Interactions
| Interaction Type | Strength | Recommended Regeneration Solutions | Alternative Solutions |
|---|---|---|---|
| Weak Acidic | pH > 2.5 | 10 mM glycine/HCl | 1-10 mM HCl |
| Intermediate Acidic | pH 2-2.5 | 10 mM Glycine/HCl | 0.5 M formic acid, 0.85% H₃PO₄ |
| Strong Acidic | pH < 2 | 10-50 mM Glycine/HCl | 1 M formic acid, 10-100 mM HCl, 0.1% trifluoracetic acid |
| Weak Basic | pH < 9 | 10 mM HEPES/NaOH | - |
| Intermediate Basic | pH 9-10 | 10 mM Glycine/NaOH | 10-100 mM NaOH |
| Strong Basic | pH > 10 | 50-100 mM NaOH | 1 M ethanolamine |
| Hydrophobic | Weak | 25-50% ethylene glycol | - |
| Hydrophobic | Intermediate | 50% ethylene glycol | 0.02% SDS |
| Hydrophobic | Strong | 25-50% ethylene glycol | 0.5% SDS |
| Ionic | Weak | 0.5-1 M NaCl | - |
| Ionic | Intermediate | 1-2 M MgCl₂, 1-2 M NaCl | - |
| Ionic | Strong | 2-4 M MgCl₂ | 6 M guanidine chloride |
Case Study: Regeneration of NTA Surfaces for His-Tagged Proteins
Specific Protocol for Cobalt(II)-NTA Chemistry
A recent advanced regeneration approach specifically designed for cobalt(II)-nitrilotriacetic acid (NTA) surfaces with His₆-tagged proteins demonstrates the complexity of modern regeneration protocols [70]. This methodology is particularly valuable for pharmaceutical applications involving tagged proteins, with the following optimized regeneration condition established for complete surface regeneration across ten cycles:
- Regeneration Buffer Composition: 100 mM EDTA, 500 mM imidazole, and 0.5% SDS at pH 8.0
- Exposure Conditions: 1 minute with shaking at 150 rpm
- Additional Treatment: Followed by washing with 0.5 M NaOH for 3 minutes
- Application: Successfully demonstrated for His₆-tagged antibody fragments, SARS-CoV-2 spike RBD protein, red fluorescent protein (RFP), and protein origami structures
Figure 2: Regeneration Protocol for Co(II)-NTA Surfaces
The key innovation in this approach is the use of Co(II)-NTA chemistry instead of the more stable Co(III)-NTA complex, which creates a sufficiently labile coordination bond with His-tags to allow regeneration while maintaining binding capacity during experimental cycles [70]. The regeneration mechanism involves multiple disruption strategies: EDTA chelates and removes cobalt ions from the NTA complex, imidazole competes with the His-tag for coordination sites, and SDS disrupts hydrophobic interactions and protein structure.
The Researcher's Toolkit: Essential Reagents and Materials
Table 3: Essential Research Reagents for SPR Regeneration Studies
| Reagent/Category | Specific Examples | Function in Regeneration |
|---|---|---|
| Acidic Buffers | 10-100 mM Glycine-HCl (pH 1.5-3.0), 0.5-1 M formic acid, 10-100 mM HCl, 0.85% H₃PO₄ | Disrupts electrostatic interactions and hydrogen bonding; causes partial protein unfolding |
| Basic Buffers | 10-100 mM NaOH, 10 mM Glycine/NaOH (pH 9-10), 1 M ethanolamine | Disrupts electrostatic interactions and hydrogen bonding |
| Chaotropic Agents | 2-6 M guanidine hydrochloride, 4-8 M urea, 1-4 M MgCl₂ | Disrupts hydrogen bonding networks; destabilizes protein structure |
| Detergents | 0.01-0.5% SDS, 0.3% CHAPS, 0.3% Zwittergent, 0.3% Tween variants | Solubilizes hydrophobic interfaces; disrupts hydrophobic interactions |
| Competitive Agents | 500 mM imidazole, ligand analogs | Competes with binding interactions for active sites |
| Chelating Agents | 100 mM EDTA, 20 mM EDTA | Removes metal ions from coordination complexes (e.g., NTA surfaces) |
| Organic Solvents | 25-50% ethylene glycol, DMSO, ethanol, acetonitrile | Reduces hydrophobic interactions; alters solvation environment |
| High Salt Solutions | 0.5-4 M NaCl, 0.46 M KSCN | Disrupts electrostatic interactions through ionic shielding |
Troubleshooting and Optimization Strategies
Diagnostic Patterns in Regeneration Challenges
Interpreting sensorgram patterns is essential for diagnosing regeneration issues:
- Progressive Baseline Rise: Indicates incomplete regeneration with analyte accumulation. Solution: Increase regeneration stringency (lower pH, add detergents, or extend exposure time).
- Progressive Baseline Decline: Signals ligand degradation or removal. Solution: Use milder regeneration conditions or shorter exposure times.
- Reduced Binding Capacity: Suggests partial ligand denaturation. Solution: Reduce regeneration buffer strength or explore alternative buffer compositions.
- Inconsistent Binding Responses: May indicate variable regeneration efficiency. Solution: Standardize regeneration contact time and flow conditions.
Advanced Optimization Techniques
For challenging regeneration scenarios, several advanced strategies can be employed:
- Multi-Step Regeneration: Sequential application of different regeneration solutions can more effectively disrupt complex binding interactions. For example, starting with a high-pH buffer followed by a specific chelating agent for metal-dependent interactions [70].
- Surface Conditioning: Performing 1-3 preliminary regeneration cycles before data collection can stabilize the ligand surface and improve reproducibility [69].
- Stabilization Periods: Incorporating buffer flow between regeneration and the next analyte injection allows surface re-equilibration and minimizes buffer Artifacts [68].
- Local Rmax Fitting: When minor ligand activity loss is unavoidable, fitting binding data to a local rather than global Rmax can accommodate slight activity variations [69].
Mastering regeneration protocols represents a critical competency in SPR research that directly impacts data quality, experimental throughput, and resource utilization. The development of effective regeneration strategies requires a systematic approach that balances complete analyte removal with preservation of ligand functionality. As demonstrated through both classical cocktail approaches and contemporary specialized protocols for specific surface chemistries, successful regeneration methodologies employ targeted disruption of the precise molecular forces governing each unique biomolecular interaction.
For researchers pursuing SPR within a comprehensive thesis framework, regeneration protocol development exemplifies the essential integration of theoretical principles with practical optimization. The techniques and troubleshooting strategies outlined in this guide provide a foundation for developing robust, reproducible regeneration protocols capable of supporting high-quality SPR research across diverse biological systems and experimental applications. As SPR technology continues to evolve toward increasingly complex biomolecular systems and higher-throughput applications, regeneration methodologies will remain an active and essential area of methodological development in biophysical analysis.
Optimizing Sample Quality and Concentration for Reliable Kinetic Data
In Surface Plasmon Resonance (SPR) research, the quality of the generated kinetic data is fundamentally dependent on the initial preparation of samples. Achieving reliable measurements of association (ka) and dissociation (kd) rate constants, leading to accurate affinity (KD) calculations, requires meticulous attention to both the quality and concentration of the interacting molecules—the ligand (immobilized partner) and the analyte (injected partner). Impurities, inappropriate concentrations, or suboptimal buffer conditions can introduce artifacts, skewing the sensorgram shapes and leading to erroneous kinetic interpretations. This guide details the foundational principles and practical protocols for optimizing these critical parameters, ensuring that the collected data truly reflects the biology of the molecular interaction under investigation.
Foundational Principles for Sample Preparation
The Critical Role of Sample Purity and Integrity
The integrity of the ligand and analyte is paramount. Impurities such as protein aggregates, denatured molecules, or contaminants can cause significant issues, including non-specific binding, signal instability, and skewed kinetic parameters [58]. Thorough purification and characterization of all samples immediately prior to the SPR experiment are essential steps to ensure that only the desired molecules are being studied [58]. For proteins, this may involve techniques like size-exclusion chromatography to remove aggregates, followed by analytical methods to confirm monodispersity and stability.
Ligand Selection and Immobilization Strategy
The first critical decision in any SPR experiment is selecting which binding partner to immobilize as the ligand. This choice should aim to simplify immobilization, maximize the signal-to-noise ratio, and minimize non-specific binding. Key factors to consider include [42]:
- Molecular Size: The smaller binding partner is generally better suited as the ligand to maximize the response signal.
- Purity: For covalent coupling strategies (e.g., amine coupling), the purest binding partner should be the ligand to ensure only the molecule of interest is attached to the surface.
- Valency: Binding partners with two or more binding sites are typically better suited as the ligand. A multivalent analyte can bind to two ligands, providing an artificially low signal.
- Tags: The presence of tags (e.g., His-tag, GST-tag) can facilitate oriented immobilization, often resulting in higher ligand activity and binding site accessibility.
Essential Reagents and Buffers
The choice of running buffer and additives directly affects the stability of the interaction, the baseline signal, and the level of non-specific binding. A well-formulated buffer maintains the integrity of the interacting species and the sensor chip surface.
Table 1: Key Research Reagent Solutions for SPR Experiments
| Reagent/Buffer | Primary Function | Key Considerations |
|---|---|---|
| Running Buffer | Maintains a stable baseline and biomolecule activity [58]. | Often contains salts for ionic strength and a detergent like Tween 20 (e.g., 0.05%) to reduce non-specific binding [4]. |
| Immobilization Buffers | Facilitates preconcentration and covalent coupling of the ligand [66]. | Low pH acetate buffers (pH 4.0-5.5) are common for amine coupling; optimal pH is ligand-dependent. |
| Regeneration Solutions | Removes bound analyte without damaging the ligand [42]. | Must be optimized for each interaction; examples include low pH (10 mM glycine-HCl, pH 2.0-3.0) or high pH (10 mM NaOH) [42] [71]. |
| Blocking Agents | Reduces non-specific binding by occupying unused active sites on the sensor surface [58]. | Ethanolamine is used after amine coupling; BSA or casein can be used as protein blockers. |
| Additives (e.g., BSA, Tween 20) | Further minimizes non-specific interactions [42]. | BSA (typically 1%) shields charged domains; Tween 20 disrupts hydrophobic interactions. |
Optimizing Analyte Concentration Series
A correctly prepared dilution series of the analyte is integral to calculating kinetics with a high degree of confidence, as both the association rate constants and the affinity constants are concentration-dependent [42].
Designing the Concentration Series
For a comprehensive kinetic analysis, a minimum of three, but ideally five, analyte concentrations are recommended. These concentrations should span a range from 0.1 to 10 times the expected KD value of the interaction [42]. This ensures the resulting sensorgrams are evenly spaced, showing clear progression from sub-saturation to full saturation (see Figure 1). If the expected KD is unknown, a preliminary experiment should be conducted, starting at a low nM concentration and increasing until a binding response is observed.
In cases where steady-state binding is reached very quickly, making full kinetic analysis difficult, an affinity (equilibrium) analysis can be performed. This requires obtaining a single data point from 8 to 10 different analyte concentrations to provide sufficient data for a plot of average response versus concentration, from which the KD can be confidently determined [42].
Quantitative Guidelines for Concentration Ranges
Table 2: Analyte Concentration Series Design for Different Analysis Types
| Analysis Type | Number of Concentrations | Recommended Range | Key Objective |
|---|---|---|---|
| Full Kinetics | 5 (minimum 3) | 0.1x to 10x KD | To capture the complete progression of the association and dissociation phases for robust ka and kd calculation [42]. |
| Affinity (Steady-State) | 8 to 10 | From zero to saturation | To obtain a reliable saturation binding curve for determining KD from the response at equilibrium [42]. |
| Pilot Experiment (Unknown KD) | 3 to 5 | Low nM, increasing incrementally | To identify a concentration range where binding is detectable but not saturating, informing a more precise series. |
To ensure accurate preparation of the concentration series, a serial dilution approach is recommended. This method minimizes pipetting errors that can accumulate when changing pipettes and volumes between each dilution step, leading to more precise and reproducible data [42].
Practical Protocols and Workflows
Preconcentration Screening for Efficient Ligand Immobilization
Preconcentration is a powerful technique for increasing the density of ligand immobilized on a carboxyl sensor chip while using minimal amounts of precious protein sample [66]. It works by adjusting the pH of the immobilization buffer to create opposite charges between the sensor surface and the ligand, causing an electrostatic accumulation of the protein on the surface prior to covalent coupling [66].
Optimization Procedure [66]:
- Prepare Ligand Solutions: Dissolve the ligand at a low concentration (5-25 μg/mL) in a series of acetate buffers (e.g., pH 4.0, 4.5, 5.0, 5.5). Ensure the ligand concentration is identical in each buffer.
- Load Sensor Chip: Place a standard Carboxyl Sensor chip into the instrument.
- Test Immobilization: For each buffer pH, perform a short injection of the ligand solution at a low flow rate (e.g., 10 μL/min).
- Regenerate Surface: Strip the non-covalently bound ligand from the surface using a regeneration solution like 10 mM HCl at a high flow rate (100 μL/min).
- Analyze Results: Plot the response curves. The optimal immobilization buffer is the one with the highest pH that produced a large signal increase, as coupling efficiency is better at higher pH.
A Workflow for Sample Quality and Concentration Optimization
The following diagram outlines a logical workflow for navigating the key steps of sample preparation and optimization, from initial design to data assessment.
Diagram 1: A logical workflow for optimizing sample quality and concentration in SPR experiments.
Troubleshooting Common Artifacts
Even with careful planning, artifacts can arise. Identifying these in the raw sensorgram data is a critical step before proceeding with data analysis [42].
Bulk Shift (Solvent Effect): This appears as a large, rapid response change at the start and end of injection, creating a "square" shape. It is caused by a difference in the refractive index between the analyte solution and the running buffer [42]. Solution: Match the components of the analyte buffer as closely as possible to the running buffer. While reference subtraction can help, it is best to minimize the effect at the source [42].
Non-Specific Binding (NSB): NSB occurs when the analyte interacts with the sensor surface itself or non-target sites on the immobilized ligand, inflating the measured response [42] [58]. Solution: Test for NSB by running a high analyte concentration over a bare sensor. Mitigation strategies include adjusting buffer pH, adding blocking agents like BSA (1%) or non-ionic surfactants like Tween 20, increasing salt concentration, or changing the sensor chemistry [42].
Mass Transport Limitation: This occurs when the diffusion of the analyte from the bulk solution to the sensor surface is slower than its association rate constant. The sensorgram will show a linear association phase with a lack of curvature [42]. Solution: Increase the flow rate, decrease the ligand density on the sensor surface, or use a higher diffusing analyte if possible [42].
In the realm of SPR research, the adage "garbage in, garbage out" holds profound truth. There is no software correction or sophisticated kinetic model that can compensate for poorly prepared samples. Optimizing sample quality through rigorous purification, selecting the appropriate ligand and immobilization strategy, and meticulously designing the analyte concentration series are not merely preliminary steps—they are the very foundation upon which reliable, publication-quality kinetic data is built. By integrating the protocols and guidelines outlined in this guide, researchers can streamline their path to robust and interpretable results, thereby solidifying the role of SPR as a cornerstone technology in fundamental research and drug discovery.
Surface Plasmon Resonance (SPR) is a powerful, label-free technology for real-time biomolecular interaction analysis (BIA), providing critical data on binding kinetics and affinity [30]. However, its sensitivity makes it particularly vulnerable to reproducibility issues, as small variations in procedure, environmental conditions, or reagent quality can significantly impact results [27]. The fundamental goal of standardizing procedures and controls is to minimize this variability, ensuring that kinetic constants such as association (ka) and dissociation (kd) rates are reliable and comparable across experiments and laboratories. This guide details the specific strategies and controls necessary to achieve this reproducibility, with a focus on practical implementation for researchers and drug development professionals.
Standardizing Core Experimental Procedures
Sensor Chip Surface Preparation
The immobilization of the ligand onto the sensor chip is a foundational step where inconsistencies can readily arise.
- Controlled Ligand Density: Immobilization levels must be optimized for the specific experimental goal. For standard kinetic analysis in a 1:1 binding model, a low density of ligand is required to avoid mass transport limitation, where the rate of analyte diffusing to the surface becomes slower than the binding rate itself [72] [30]. Conversely, Calibration-Free Concentration Analysis (CFCA) intentionally uses a high ligand density to create a partially mass-transport limited system for direct measurement of active analyte concentration [72].
- Immobilization Chemistry: The chosen chemistry must provide a stable and consistent covalent attachment. Common approaches include amine coupling using carboxymethylated dextran chips (e.g., CM5) [30]. For sensitive targets like G Protein-Coupled Receptors (GPCRs), maintaining stability requires advanced strategies such as immobilization within native membranes, liposomes, nanodiscs, or through engineered tags to ensure the receptor remains functional [5].
- Regeneration Scouting: A rigorous and standardized regeneration procedure is essential for re-using the sensor chip surface without damaging the immobilized ligand. This involves testing various regeneration solutions (e.g., low pH, high salt) to identify the condition that completely dissociates the bound analyte while preserving ligand activity for subsequent cycles [30].
Analytic Preparation and Characterization
Inconsistencies in the analyte are a major source of irreproducibility.
- Determining Active Concentration: Traditional methods like absorbance at 280 nm measure total protein concentration but fail to distinguish the functionally active fraction from misfolded or denatured protein [72]. This can lead to significant errors in calculated binding constants. Employing CFCA via SPR or other methods that specifically quantify the active concentration of the protein reagent is critical for accurate and reproducible results [72].
- Sample Purity and Buffer Composition: Analyte samples should be of high and consistent purity. The running buffer composition (pH, ionic strength, additives) must be meticulously controlled, as these factors can profoundly influence binding kinetics and stability.
Data Collection and Flow Parameters
Standardizing the instrument operation itself is key to obtaining comparable sensorgrams.
- Flow Rate Control: A constant, sufficiently high flow rate is recommended for most kinetic analyses to minimize mass transport effects. Varying the flow rate is also a useful diagnostic tool to check for mass transport limitation [27] [30]. The binding data for CFCA requires analyte to be run over the surface at two different flow rates to determine the active concentration [72].
- Temperature Stabilization: SPR measurements are sensitive to temperature fluctuations. Instruments should be allowed to equilibrate, and the temperature should be actively controlled and documented for all experiments. Lack of temperature control is a noted source of variance in real-world measurements [27].
Table 1: Key Experimental Parameters for Standardization
| Parameter | Standardization Goal | Impact on Reproducibility |
|---|---|---|
| Ligand Density | Optimized for the specific assay (low for kinetics, high for CFCA) | Prevents mass transport artifacts; ensures accurate kinetic and concentration data [72] [30]. |
| Active Analyte Concentration | Use of CFCA or equivalent methods | Provides correct input for affinity/kinetic calculations; eliminates variability from protein quality [72]. |
| Flow Rate | Constant and sufficiently high; dual rates for CFCA | Ensines consistent analyte delivery; used to calculate concentration in CFCA [72] [30]. |
| Regeneration | A validated, consistent protocol | Allows for multiple reliable analyte injections on the same ligand surface [30]. |
| Temperature | Actively controlled and monitored | Prevents drift in baseline and binding responses caused by thermal noise [27]. |
Implementing Rigorous Environmental and Reagent Controls
Critical Reagent Quality and Characterization
Protein-based reagents are a recognized source of variability in bioanalysis. The poor quality of commercial reagents has been estimated to cause hundreds of millions of dollars in annual waste due to irreproducible research [72]. Key quality attributes include:
- Concentration: Must be designated and verified using active concentration methods [72].
- Purity and Stability: Assessed via techniques like SDS-PAGE and size-exclusion chromatography. However, purity analysis alone offers little insight into the level of active protein present [72].
- Specificity and Function: Must be validated for the intended assay.
Table 2: Scientist's Toolkit - Essential Research Reagent Solutions
| Reagent / Material | Function in SPR Experiments |
|---|---|
| Sensor Chips (e.g., CM5, C1, NTA, L1) | Solid support with a gold film and various surface chemistries for ligand immobilization. Choice depends on ligand properties and assay needs (e.g., L1 for capturing liposomes) [30]. |
| Coupling Reagents (NHS/EDC) | Activates carboxylated dextran surfaces for covalent amine coupling of ligands [30]. |
| Regeneration Solutions (e.g., low pH, high salt) | Dissociates bound analyte after each injection to regenerate the ligand surface for the next cycle [30]. |
| Running Buffers (e.g., PBS, HBS-EP) | Maintain a constant chemical environment; HBS-EP includes additives to reduce non-specific binding. |
| Capture Ligands (e.g., Streptavidin, anti-tag antibodies) | Used in capture coupling methods to immobilize biotinylated or tagged ligands in a uniform orientation. |
Data Evaluation and Model Selection
The evaluation of sensorgram data is a critical control point often overlooked.
- Avoiding "Black Box" Software: Relying solely on commercial software without understanding the underlying models can lead to the application of incorrect kinetics and false constants [27].
- Model Validation: The assumed binding model (e.g., 1:1 interaction) must be checked for its correctness. This requires an understanding of both the biomolecular processes and the kinetic processes at the biosensor surface [27] [30]. Data should be fitted using global fitting across multiple analyte concentrations rather than just a single curve.
- Awareness of Artefacts: Researchers must be able to identify and account for potential artefacts such as mass transport limitation, non-specific binding, or analyte aggregation [27].
SPR Standardized Workflow
A Practical Protocol for Reproducible SPR Analysis
The following integrated protocol synthesizes the above controls into a actionable workflow. This protocol is adapted from established methodologies [30] with an emphasis on steps that ensure reproducibility.
- Ligand and Analyte Preparation: Express and purify both interaction partners. For the analyte, determine the active concentration using a method like CFCA, not just A280. For the ligand, confirm functionality.
- Sensor Chip Selection: Choose a chip based on your ligand and assay. Common choices are CM5 for general amine coupling, SA for biotinylated ligands, or L1 for membrane-based assays [30].
- Immobilization Optimization: Dilute the ligand in buffers at different pHs to determine the condition for optimal surface concentration. Immobilize the ligand to the desired density (Rmax), documenting all parameters.
- Regeneration Scouting: Inject a high concentration of analyte followed by various regeneration solutions. Identify the mildest condition that returns the signal to baseline without damaging the ligand surface.
- Binding Experiment: Using a concentration series of analyte (ideally spanning 10x below to 10x above the expected Kd), inject samples over the immobilized ligand surface. Maintain a constant, controlled temperature and a flow rate high enough to minimize mass transport.
- Data Analysis: Fit the collective sensorgram data to an appropriate kinetic model using global fitting. Validate the model by checking the goodness of fit and residual patterns.
SPR Reproducibility Control Framework
Achieving reproducibility in SPR research is not a single action but the result of a comprehensive strategy addressing all phases of experimentation. It requires moving beyond basic protocols to implement strict standardization of procedures, rigorous control of reagents and environment, and a critical, informed approach to data evaluation. By focusing on the accurate determination of active reagent concentration, controlling ligand density and flow dynamics, and validating kinetic models, researchers can generate reliable, high-quality data. This discipline is fundamental for advancing drug discovery, particularly for challenging targets like GPCRs, and for building a trustworthy foundation of biomolecular interaction knowledge.
Advancements and Validation: Enhancing SPR Sensitivity and Comparative Analysis
Surface Plasmon Resonance (SPR) is a cornerstone optical technique for real-time, label-free detection of biomolecular interactions. The fundamental principle relies on the excitation of surface plasmon polaritons—collective oscillations of free electrons at a metal-dielectric interface [6]. When polarized light strikes a metal film under specific conditions, a resonance energy transfer occurs, which is extremely sensitive to minute changes in the refractive index at the sensor surface [73] [74]. This enables SPR sensors to monitor binding events, such as antigen-antibody interactions, with exceptional precision. The performance of an SPR biosensor is quantified by key parameters including sensitivity (the resonance shift per unit refractive index change), figure of merit (FOM) (the ratio of sensitivity to resonance width), and detection accuracy [75] [74].
While traditional SPR sensors utilize thin films of noble metals like gold and silver, their performance is constrained by inherent limitations in signal strength and biomolecular affinity. Next-generation sensing materials are engineered to overcome these barriers by enhancing the local electromagnetic field, improving chemical stability, and providing greater surface area for analyte capture [73]. The integration of advanced materials such as two-dimensional (2D) nanomaterials, metal oxides, and bimetallic structures has propelled SPR technology toward unprecedented levels of sensitivity and specificity, unlocking new potentials in medical diagnostics, environmental monitoring, and drug development [6] [76].
Two-Dimensional Materials for SPR Enhancement
Properties and Enhancement Mechanisms
Two-dimensional materials, characterized by their atomic-scale thickness and extensive lateral dimensions, have emerged as powerful enhancers for SPR biosensors. Their exceptional properties, including ultra-high surface-to-volume ratios, exceptional charge carrier mobility, and strong light-matter interactions, make them ideal for amplifying plasmonic signals [73] [77]. When layered onto a conventional metallic SPR film, these materials act as molecular adsorption layers, significantly concentrating analyte molecules within the enhanced evanescent field. This effectively increases the local refractive index change upon binding, leading to a more pronounced resonance shift [73]. Furthermore, many 2D materials possess functional groups or tunable surface chemistries that facilitate robust immobilization of biorecognition elements such as antibodies and aptamers [74] [78].
Material Classes and Performance
The family of 2D nanomaterials is diverse, with each member offering unique advantages for SPR sensing.
Graphene and Graphene Oxide (GO): Graphene's gapless linear dispersion and biocompatibility make it a premier material for enhancing SPR sensors. It protects metal films from oxidation and increases the adsorption of analyte molecules via π-π stacking interactions [73] [78]. Graphene oxide, with its oxygen-containing functional groups, allows for easy functionalization and has been shown to significantly boost sensitivity. For instance, a sensor incorporating GO and silver-gold bimetallic nanoparticles demonstrated a refractive index sensitivity of 4715.9 nm/RIU [78].
MXenes: MXenes, such as Ti₃C₂Tₓ, are a class of 2D transition metal carbides/nitrides known for their high metallic conductivity and hydrophilic surfaces. Their integration into SPR sensors leads to a substantial performance leap. A Kretschmann-configured sensor with layers of Au/graphene/Al₂O₃/MXene achieved an exceptional angular sensitivity of 163.63 °/RIU and a FOM of 17.52 RIU⁻¹, making it a cutting-edge tool for detecting cancer biomarkers like carcinoembryonic antigen (CEA) [74].
Transition Metal Dichalcogenides (TMDs): Materials like MoS₂ exhibit strong excitonic effects and high absorption coefficients. When used in van der Waals heterojunctions with graphene, they significantly improve electric field enhancement at the sensing interface, leading to highly sensitive platforms for gas molecule and biomolecule detection [73] [77].
Table 1: Performance of SPR Sensors Enhanced with 2D Materials
| 2D Material | Sensor Configuration | Sensitivity | Figure of Merit (FOM) | Key Application |
|---|---|---|---|---|
| MXene (Ti₃C₂Tₓ) | BK7 prism/Au/Graphene/Al₂O₃/MXene [74] | 163.63 °/RIU | 17.52 RIU⁻¹ | Carcinoembryonic Antigen (CEA) detection |
| Graphene Oxide (GO) | Ag@Au/GO optical fiber [78] | 4715.9 nm/RIU | - | Human IgG detection |
| MoS₂/Graphene | Van der Waals heterojunction [77] | Enhanced electric field | - | Gas molecule detection |
Experimental Protocol: Fabrication of a 2D Material-Enhanced SPR Sensor
The following workflow details the construction of an SPR sensor integrated with MXene and graphene for ultra-sensitive biomarker detection, based on a Kretschmann configuration [74].
Title: SPR Sensor Fabrication Workflow
Detailed Steps:
- Prism Preparation: Begin with a clean BK7 prism. Standard cleaning procedures (e.g., piranha solution, oxygen plasma) are crucial to ensure a contaminant-free surface for uniform layer deposition [74].
- Metal Deposition: Deposit a ~50 nm thick gold film onto the prism using a physical vapor deposition technique such as magnetron sputtering or thermal evaporation. The gold film serves as the primary plasmonic layer [74].
- 2D Material Transfer: Transfer a single layer of graphene onto the gold film using a wet-transfer or dry-transfer method like PMMA-assisted transfer. This layer enhances the evanescent field and provides a protective barrier [74].
- Oxide Layer Deposition: Deposit a thin layer of aluminum oxide (Al₂O₃) via atomic layer deposition (ALD) to a precise thickness (e.g., 5-10 nm). This dielectric layer acts as a spacer to fine-tune the resonance conditions and improve detection accuracy [74].
- MXene Coating: Prepare an aqueous dispersion of MXene (Ti₃C₂Tₓ) and spin-coat it onto the Al₂O₃ layer. The thickness can be controlled by the concentration of the dispersion and the spin speed. MXene's high surface area and functional groups are key to signal amplification [74].
- Functionalization: Immobilize specific biorecognition elements, such as antibodies or aptamers, onto the MXene surface. This is often achieved using carbodiimide crosslinking chemistry (EDC/NHS) to form amide bonds with the functional groups on the 2D material [74] [78].
- Sensor Assembly and Characterization: Integrate the fabricated sensor chip into a flow cell system. Connect to a microfluidic pump and a high-resolution spectrometer. Characterize the sensor's performance by measuring its response to solutions of known refractive index (e.g., glycerol solutions) to calculate sensitivity and FOM [74].
Metal Oxides as Sensitivity Boosters
Role and Function in SPR Sensors
Metal oxides represent a class of high-refractive-index dielectric materials that significantly boost SPR sensor performance by intensifying the evanescent field at the sensing interface. Their primary mechanism of action is to increase the equivalent refractive index of the analyte medium, which leads to a more pronounced shift in the resonance condition upon molecular binding [79]. This makes the sensor more responsive to minute changes in the local environment. Furthermore, some metal oxides, such as TiO₂ and ZnO, exhibit excellent chemical stability, high surface-to-volume ratios, and a strong affinity for biomolecules, making them ideal coating materials for enhancing both the sensitivity and robustness of fiber-optic SPR sensors [79] [73].
Comparative Performance of TiO₂ and ZnO
Experimental studies have directly compared the enhancement effects of different metal oxides. In one investigation, a silver-based optical fiber sensor was coated with TiO₂ and ZnO films of identical thickness. The results demonstrated that the metal oxide with the higher effective refractive index provided a greater improvement in sensitivity [79].
- TiO₂ (Titanium Dioxide): Exhibited a maximum refractive index sensitivity of 15,200 nm/RIU, a 16.92% increase compared to a traditional silver-based sensor.
- ZnO (Zinc Oxide): Showed a sensitivity of 13,600 nm/RIU, which is a 4.62% improvement over the baseline sensor [79].
This confirms that the selection of metal oxide material is a critical design parameter, with TiO₂ offering superior enhancement in this specific configuration.
Table 2: Performance Comparison of Metal Oxide-Enhanced SPR Sensors
| Metal Oxide | Sensor Configuration | Refractive Index Sensitivity | Enhancement vs. Ag-only | Other Applications |
|---|---|---|---|---|
| Titanium Dioxide (TiO₂) | Ag-based fiber sensor with TiO₂ coating [79] | 15,200 nm/RIU | +16.92% | Temperature sensing (4.9 nm/°C) |
| Zinc Oxide (ZnO) | Ag-based fiber sensor with ZnO coating [79] | 13,600 nm/RIU | +4.62% | Temperature sensing (4.3 nm/°C) |
Bimetallic Layers and Alloyed Structures
Advantages Over Single-Metal Films
Bimetallic configurations in SPR sensors combine two different metals to synergize their individual advantages and mitigate their weaknesses. A common challenge with single-metal films is the trade-off between performance and practicality; for instance, silver supports sharp resonances and high sensitivity but is prone to oxidation, while gold is chemically inert but generally yields a broader resonance and lower sensitivity [80]. Bimetallic layers, such as silver-gold (Ag-Au) and copper-gold (Cu-Au), are engineered to harness the superior plasmonic properties of one metal while using the other to provide stability and functionalization capabilities. For example, a structure with a silver core and a thin gold shell (Ag@Au) leverages the strong plasmonic response of silver while the gold shell prevents oxidation and facilitates easy biomolecule immobilization [78].
Performance of Bimetallic Configurations
Research into various multilayer topologies has revealed significant performance gains. A study on D-shaped optical fiber sensors investigated numerous combinations of Au, Ag, and Cu with graphene.
- The Cu/Ag bimetallic pairing, prior to the addition of other enhancing materials, achieved a sensitivity of 147 °/RIU. This represents a remarkable 390% improvement over a single silver layer and a 267.5% improvement over a single copper layer [75].
- Certain configurations, including Ag-Au and Ag-C-Au, demonstrated a maximum wavelength sensitivity of 70,000 nm/RIU [80].
- The integration of bimetallic nanoparticles with 2D materials yields even greater performance. The Ag@Au/GO fiber SPR sensor achieved a refractive index sensitivity of 4715.9 nm/RIU and was successfully applied to detect human IgG with a high immunoassay sensitivity [78].
Emerging Composites: Halide Perovskites with Bimetallics
A cutting-edge advancement involves integrating metal halide perovskites with bimetallic structures. These perovskites, known for their excellent optical properties and tunability, further enhance light-matter interactions. When an 8 nm layer of Cesium Tin Iodide (CsSnI₃) was added to a Cu/Ag bimetallic structure, the sensitivity skyrocketed to 460 °/RIU, showcasing the immense potential of such hybrid material systems [75].
Table 3: Performance of Bimetallic and Composite SPR Sensors
| Material Structure | Sensor Configuration | Sensitivity | Key Advantage |
|---|---|---|---|
| Ag@Au/GO | Optical fiber with Ag-core, Au-shell, and GO [78] | 4715.9 nm/RIU (RI), 0.53 nm/μg/mL (IgG) | Prevents Ag oxidation, high immunoassay sensitivity |
| Cu/Ag Bimetallic | Kretschmann configuration [75] | 147 °/RIU | Cost-effective, high performance vs. single metals |
| Cu/Ag/CsSnI₃ | Kretschmann with perovskite [75] | 460 °/RIU | Enhanced light-matter interaction |
| Various Multilayers | D-shaped optical fiber (e.g., Ag-Au, Ag-C-Au) [80] | 70,000 nm/RIU (Wavelength) | Ultra-high wavelength sensitivity |
The Scientist's Toolkit: Essential Research Reagents and Materials
Successful development and fabrication of next-generation SPR sensors require a suite of specialized materials and reagents. The following table details the core components of the research toolkit.
Table 4: Essential Research Reagent Solutions for SPR Sensor Development
| Reagent/Material | Function/Application | Specific Example |
|---|---|---|
| Gold (Au) & Silver (Ag) Targets | Physical vapor deposition (sputtering/evaporation) of plasmonic metal films. | High-purity (99.99%) Au and Ag targets for film deposition [74]. |
| 2D Material Dispersions | Coating of metal films to enhance evanescent field and provide functional groups. | Aqueous dispersions of Graphene Oxide (GO), MXene (Ti₃C₂Tₓ) [74] [78]. |
| Crosslinking Agents | Activating carboxyl groups on 2D materials for covalent immobilization of biomolecules. | EDC (1-Ethyl-3-(3-dimethylaminopropyl)carbodiimide) and NHS (N-Hydroxysuccinimide) mixture [78]. |
| Biorecognition Elements | Providing specificity for the target analyte. | Antibodies (e.g., Rabbit anti-human IgG), Staphylococcal Protein A (SPA) for oriented antibody immobilization [78]. |
| Blocking Agents | Passivating unused active sites on the sensor surface to minimize non-specific binding. | Bovine Serum Albumin (BSA) or ethanolamine solution [78]. |
| Regeneration Solutions | Breaking antigen-antibody bonds to regenerate the sensor surface for reuse. | Low-pH buffers (e.g., 10 mM Glycine-HCl) or mild alkaline solutions (e.g., 10 mM NaOH) [78]. |
The integration of two-dimensional materials, metal oxides, and bimetallic layers represents a paradigm shift in the design of Surface Plasmon Resonance sensors. These advanced materials directly address the limitations of conventional single-metal films, leading to transformative improvements in sensitivity, stability, and functional versatility. As research progresses, the focus is expanding toward the development of sophisticated hybrid structures that combine the strengths of multiple material classes, such as perovskites with bimetallics or TMDs with graphene [75] [77].
Future advancements will be driven by the synergy between material science and computational approaches. The integration of machine learning and artificial intelligence is poised to play a pivotal role in optimizing complex sensor designs, analyzing sensor array data, and improving predictive modeling for multi-analyte detection [76]. While challenges in scalable fabrication and cost-effective manufacturing remain, the ongoing innovation in next-generation sensing materials firmly establishes SPR technology as an increasingly powerful and indispensable tool for researchers and drug development professionals pushing the boundaries of analytical science.
Surface Plasmon Resonance (SPR) biosensing represents a cornerstone technology in label-free optical biosensing, enabling the real-time investigation of biomolecular interactions through the measurement of refractive index changes at a sensor surface [81]. The fundamental principle underpinning SPR research involves the excitation of surface plasmon polaritons at a metal-dielectric interface, typically a thin gold or silver film, which is sensitive to minute changes in the local refractive index caused by binding events [6]. This technical case study examines the validation of a novel, high-sensitivity SPR biosensor engineered specifically for the detection of Mycobacterium tuberculosis, positioning this development within the broader context of advancing SPR fundamentals for medical diagnostics. Tuberculosis remains a leading global health challenge, necessitating diagnostic technologies that offer rapid, accurate, and sensitive detection to facilitate timely treatment and curb disease transmission [53]. The biosensor discussed herein addresses these requirements through a sophisticated multilayered architecture and rigorous experimental validation, demonstrating significant performance enhancements over previously reported SPR configurations.
Biosensor Design and Theoretical Foundation
Multilayer Architecture and Materials Selection
The proposed SPR biosensor employs a meticulously engineered multilayered configuration structured as CaF₂/SiO₂/Ag/AlON/BP [53]. This design integrates a calcium fluoride (CaF₂) prism as the coupling component, followed by sequential layers of silicon dioxide (SiO₂), silver (Ag), aluminium oxynitride (AlON), and black phosphorus (BP). The selection of each material is grounded in its specific optical and functional properties that collectively enhance the plasmonic response. The CaF₂ prism provides a high refractive index substrate for efficient photon-to-plasmon conversion, while the silver layer serves as the primary plasmonic material due to its excellent optical properties in the visible and near-infrared spectrum. The incorporation of two-dimensional black phosphorus (BP) and AlON as functional layers significantly enhances the biological interaction capabilities and overall sensor stability [53].
An alternative optimized design utilizing a CaF₂TiO₂/Ag/TiO₂/black phosphorus configuration has also been reported, demonstrating the continuous innovation in material stacks for tuberculosis detection [82]. This configuration leverages the differential evolution (DE) algorithm for structural parameter optimization, representing an advanced computational approach to sensor design that distinguishes it from conventional iterative methods [82].
Performance Metrics and Theoretical Validation
The theoretical validation of the biosensor was conducted through comprehensive numerical simulations employing the Transfer Matrix Method (TMM) and Finite Element Method (FEM) [53]. These computational techniques are fundamental to SPR research for modeling light-matter interactions and electromagnetic field distribution in complex multilayered structures. The TMM approach provides an analytical solution for reflectance calculations based on the stratification of optical media, while FEM enables more granular analysis of field enhancements and localized effects at the sensor interface.
The proposed biosensor achieves a remarkable angular sensitivity of 615.33 deg/RIU [53], with an alternative design further pushing this parameter to 654 deg/RIU through DE algorithm optimization [82]. This sensitivity quantifies the angular shift in the resonance dip per unit change in refractive index (RIU), representing a crucial figure of merit for SPR biosensors. Additional performance parameters include a quality factor (QF) of 275.29 RIU⁻¹, detection accuracy (DA) of 0.50 deg⁻¹, and figure of merit (FOM) of 1206.3 RIU⁻¹ [53]. These metrics collectively demonstrate the sensor's capability for precise detection within the biological refractive index range of 1.29 to 1.35, encompassing most clinically relevant analytes including bacterial cells and biomarkers [53].
Table 1: Key Performance Metrics of the High-Sensitivity SPR Biosensor
| Performance Parameter | Value | Significance |
|---|---|---|
| Angular Sensitivity | 615.33 - 654 deg/RIU | Measures angular shift per refractive index unit change; higher values enable detection of lower analyte concentrations |
| Quality Factor (QF) | 275.29 RIU⁻¹ | Indicates sharpness of resonance peak; higher values improve detection resolution |
| Detection Accuracy (DA) | 0.50 deg⁻¹ | Defines precision in determining resonance angle position |
| Figure of Merit (FOM) | 1206.3 RIU⁻¹ | Comprehensive performance indicator combining sensitivity and resonance width |
| Full Width at Half Maximum (FWHM) | 2.66° | Narrow resonance width enables more accurate tracking of binding events |
| Refractive Index Detection Range | 1.29 - 1.35 | Covers biological analytes including proteins, antibodies, and whole bacterial cells |
The following diagram illustrates the fundamental architecture and working principle of the multilayered SPR biosensor:
Diagram 1: Fundamental architecture and working principle of the multilayered SPR biosensor for Mycobacterium tuberculosis detection. The diagram shows the sequential layers and light path through the system.
Experimental Protocols and Methodologies
Sensor Fabrication and Functionalization
The experimental realization of the SPR biosensor requires precise fabrication and functionalization protocols to translate the theoretical design into a functional diagnostic device. For tuberculosis detection, both antibody-antigen and DNA hybridization approaches have been successfully implemented in SPR platforms.
In an array-based SPR biosensor developed for TB antibody detection, the sensor surface was functionalized with nine different Mycobacterium tuberculosis antigens, including secreted proteins (W06, W10, W28, W64, W70), heat shock proteins (W14), lipoproteins (W19, W38), and fibronectin-binding protein (W85) [83]. The immobilization protocol involved forming a self-assembled monolayer (SAM) of 8-mercaptooctanoic acid (8-MOA) on the gold sensor surface, followed by activation with EDC/NHS (1-ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride/N-hydroxysuccinimide) chemistry to create amine-reactive esters. The antigens were then covalently attached to the activated surface at a concentration of 50 μg/mL, with subsequent blocking using 1M ethanolamine to minimize non-specific binding [83].
For nucleic acid-based detection, thiolated probe single-strand oligodeoxynucleotides (ssODNs) complementary to target sequences of Mycobacterium tuberculosis complex were immobilized on gold sensor surfaces using 6-mercapto-1-hexanol as an orientation helper and surface blocking agent [54]. This approach enables specific detection of TB DNA through hybridization events, with the sensor platform demonstrating a detection limit of 30 ng/μL and the ability to detect specific DNA hybridization at a concentration of 0.05 μM [54].
Sample Analysis and Binding Measurement
Serum samples from TB patients and healthy controls were analyzed using the array-based SPR biosensor with a flow rate of 30 μL/min [83]. The SPR measurements were conducted using a fixed angle slightly smaller than the SPR angle, with images collected using a CCD camera at 790 nm. The measured response values were expressed as ΔR, representing the normalized reflectivity change. Real-time SPR binding curves were monitored during sample injection, with serum from TB patients producing significantly higher reflectivity responses (R > 0.6) compared to healthy donors (R = 0.2-0.5) [83].
To ensure data reliability, several quality control measures were implemented. Non-specific binding tests were performed by running high analyte concentrations over sensors with no immobilized ligand [48]. Bulk refractive index effects were minimized by ensuring matching running and analyte buffers. For kinetic analysis, multiple analyte concentrations were tested in duplicate or triplicate to obtain standard deviations and ensure measurement consistency [48].
Table 2: Key Research Reagent Solutions for SPR Biosensor Implementation
| Reagent/Chemical | Function/Application | Experimental Role |
|---|---|---|
| 8-Mercaptooctanoic acid (8-MOA) | Self-assembled monolayer formation | Creates functionalized surface for biomolecule immobilization via thiol-gold chemistry |
| EDC/NHS | Carboxyl group activation | Forms amine-reactive esters for covalent coupling of proteins to the sensor surface |
| 6-Mercapto-1-hexanol | Surface blocking agent | Prevents non-specific binding and improves orientation of immobilized biomolecules |
| Ethanolamine | Surface blocking | Deactivates remaining reactive groups after immobilization to minimize non-specific binding |
| TB Antigens (W06, W10, W14, etc.) | Recognition elements | Specifically capture anti-TB antibodies from patient samples through antibody-antigen interaction |
| Thiolated ssODN Probes | DNA hybridization detection | Complementary sequences for specific detection of Mycobacterium tuberculosis DNA targets |
| HCl Regeneration Solution | Surface regeneration | Removes bound analytes while preserving immobilized ligands for sensor chip reuse |
The experimental workflow for validating the SPR biosensor, from surface functionalization to data analysis, is illustrated below:
Diagram 2: Experimental workflow for SPR biosensor validation, showing key steps from surface functionalization to data analysis and surface regeneration.
Results and Diagnostic Performance
Analytical Sensitivity and Specificity
The validated SPR biosensor demonstrated exceptional analytical performance for tuberculosis detection. In the array-based configuration targeting multiple TB antibodies, the biosensor distinguished serum specimens from TB patients and healthy controls with high reliability [83]. The real-time SPR binding curves showed significantly different response patterns, with TB patient serum inducing substantial reflectivity changes (R > 0.6) at specific antigen spots, while healthy donor samples produced minimal responses (R = 0.2-0.5) [83].
Statistical analysis established a threshold value of 0.58 (mean + twofold standard deviation from healthy group) to classify samples as positive or negative for TB infection [83]. Using this cutoff, the SPR-based sensor achieved specificity above 90% (93-100%) with sensitivity ranging from 14-79% depending on the specific antigen [83]. These performance metrics highlight the robust discriminatory power of the biosensor while indicating potential for further optimization to enhance sensitivity.
For DNA-based detection, the SPR biosensor demonstrated a detection limit of 30 ng/μL for target DNA, with the ability to detect specific hybridization at concentrations as low as 0.05 μM [54]. The sensor platform maintained functionality after regeneration with 2.5 mM HCl and could be reused multiple times without significant signal degradation. Furthermore, the sensor chips retained stability when stored in vacuum at room temperature for up to 12 weeks, indicating excellent shelf-life for practical applications [54].
Comparative Analysis with Conventional Methods
When compared to conventional diagnostic methods, the SPR biosensor offers significant advantages. Traditional smear microscopy, while simple and inexpensive, suffers from poor sensitivity [83]. Culture methods, considered the gold standard, require 6-8 weeks to provide results due to the slow-growing nature of mycobacteria, delaying critical treatment decisions [83]. Nucleic acid amplification tests like PCR, while sensitive, require complex sample purification and sophisticated instrumentation [83].
The SPR biosensor demonstrated comparable specificity to commercial ELISA tests while offering the additional advantages of label-free detection, real-time monitoring, and multiplexing capability [83]. The array format enables simultaneous detection of multiple antibodies, providing a more comprehensive serological profile than single-analyte tests. Furthermore, the rapid analysis time (minutes to hours) represents a significant improvement over culture methods and even some molecular techniques that require batch processing [83].
This case study validates a high-sensitivity SPR biosensor for Mycobacterium tuberculosis detection, demonstrating significant advancements in both design methodology and diagnostic performance. The integration of novel materials including black phosphorus and AlON in a optimized multilayer architecture, combined with sophisticated computational design using the differential evolution algorithm, has yielded a biosensor with exceptional angular sensitivity (615.33-654 deg/RIU) and comprehensive performance metrics [53] [82].
Within the broader context of SPR research fundamentals, this work exemplifies the ongoing evolution from conventional prism-based configurations toward sophisticated nanophotonic structures engineered for specific diagnostic applications. The implementation of both antibody-antigen and DNA hybridization assays on SPR platforms highlights the versatility of this technology for tuberculosis detection, offering pathways to address different stages of infection and disease manifestations [83] [54].
The validation of this biosensor contributes fundamentally to SPR research by demonstrating the practical application of advanced material stacks, computational optimization algorithms, and multiplexed detection schemes. Future research directions include further miniaturization for point-of-care applications, integration with microfluidic sample handling systems, and expansion of multiplexing capabilities to enhance diagnostic accuracy through simultaneous detection of multiple biomarkers. The continued refinement of SPR biosensors for tuberculosis diagnosis holds significant promise for addressing critical gaps in global TB control efforts, potentially enabling earlier detection, more effective treatment monitoring, and ultimately reduced transmission of this persistent pathogen.
Surface Plasmon Resonance (SPR) research has fundamentally transformed biochemical sensing by enabling label-free, real-time monitoring of molecular interactions. The fundamental principle underpinning SPR sensors involves the excitation of surface plasmon polaritons—coherent electron oscillations at the metal-dielectric interface—which generate an exponentially decaying evanescent field exquisitely sensitive to refractive index changes within approximately 200-300 nanometers of the sensing surface [84] [85]. This phenomenon provides the physical basis for detecting biological binding events without labels. However, despite their widespread adoption in life sciences, drug discovery, and medical diagnostics, traditional SPR sensor designs often face limitations in detection sensitivity, accuracy, and resolution, particularly for low-molecular-weight analytes and low-concentration samples [86] [87].
The intricate multilayer architecture of modern SPR biosensors presents a complex optimization challenge where conventional parameter scanning methods prove inadequate. Fixed-parameter scanning (FPS), which optimizes one variable while holding others constant, is notoriously time-consuming and often fails to identify globally optimal configurations due to its inability to account for synergistic interactions between structural parameters [85] [88]. This limitation has prompted researchers to explore computational intelligence algorithms capable of navigating complex, high-dimensional design spaces. Among these approaches, Differential Evolution (DE) has emerged as a particularly powerful optimization strategy for SPR sensor design, demonstrating superior performance in achieving heightened sensitivity, broader detection ranges, and lower limits of detection compared to traditional methods [82] [89] [88].
This technical guide examines the fundamental principles, implementation methodologies, and performance outcomes of DE-based optimization in SPR sensor design, with particular emphasis on its applications in biomedical detection and environmental monitoring. By framing this discussion within the broader context of SPR research fundamentals, we aim to provide researchers and sensor designers with comprehensive insights into this cutting-edge approach to sensor enhancement.
Fundamentals of Surface Plasmon Resonance Sensing
Physical Principles and Configurations
Surface Plasmon Resonance occurs when incident light photons couple with free electron oscillations at a metal-dielectric interface under specific resonance conditions. The fundamental requirement for SPR excitation is the matching of wave vectors between the incident light and surface plasmon wave, mathematically expressed as:
[ kx = \frac{2\pi}{\lambda} np \sin\theta = \frac{2\pi}{\lambda} \operatorname{Re} \left{ \sqrt{\frac{\varepsilonm \varepsilond}{\varepsilonm + \varepsilond}} \right} = k_{spp} ]
where (kx) represents the component of the incident light wave vector parallel to the interface, (\lambda) is the wavelength of incident light, (np) is the refractive index of the prism, (\theta) is the angle of incidence, (\varepsilonm) and (\varepsilond) are the dielectric constants of the metal and dielectric medium, respectively, and (k_{spp}) denotes the surface plasmon wave vector [84]. This resonance condition manifests as a sharp dip in reflectance at a specific incident angle or wavelength, with the precise resonance point shifting in response to refractive index changes within the evanescent field region.
The most prevalent configuration for exciting SPR employs the Kretschmann prism coupler, where a thin metal film (typically gold or silver) is deposited directly onto the prism base, and p-polarized light undergoes total internal reflection, generating an evanescent wave that penetrates the metal layer to excite surface plasmons at the metal-dielectric interface [84]. Alternative configurations include grating-coupled SPR and waveguide-based approaches, each with distinct advantages for specific applications. Recent advancements have incorporated two-dimensional (2D) materials such as graphene, MXene, and black phosphorus into the sensing architecture to enhance electric field strength and provide additional biomolecular adsorption sites, thereby significantly improving sensitivity [85] [88].
Key Performance Metrics in SPR Sensing
The effectiveness of SPR biosensors is quantified through several critical performance parameters that must be balanced during the optimization process:
- Sensitivity (S): Defined as the shift in resonance signal (angle, wavelength, or phase) per unit change in refractive index (RI), typically expressed as deg/RIU (degrees per refractive index unit) or nm/RIU (nanometers per refractive index unit). This represents the primary metric for evaluating sensor response to analyte binding [82] [85].
- Figure of Merit (FOM): Calculated as sensitivity divided by the full width at half maximum (FWHM) of the resonance curve, providing a combined measure of sensitivity and resonance sharpness that indicates overall detection capability [87] [86].
- Detection Accuracy (DA): Influenced by the signal-to-noise ratio (SNR) and resonance depth, determining the precision with which the resonance minimum can be determined [82] [90].
- Limit of Detection (LOD): The lowest analyte concentration that can be reliably detected, often expressed in molar concentration (M) or mass per volume (g/mL) [87] [91].
Table 1: Key Performance Metrics for SPR Biosensors
| Metric | Definition | Formula | Preferred Value |
|---|---|---|---|
| Angular Sensitivity | Resonance angle shift per RIU change | (\Delta\theta/\Delta n) (deg/RIU) | Higher |
| Wavelength Sensitivity | Resonance wavelength shift per RIU change | (\Delta\lambda/\Delta n) (nm/RIU) | Higher |
| Phase Sensitivity | Phase shift per RIU change | (\Delta\phi/\Delta n) (deg/RIU) | Higher |
| Full Width at Half Maximum (FWHM) | Angular/wavelength width of resonance curve at half depth | - | Narrower |
| Figure of Merit (FOM) | Ratio of sensitivity to resonance width | (S/FWHM) (1/RIU) | Higher |
| Detection Limit | Minimum detectable analyte concentration | - | Lower |
Differential Evolution Algorithm: Principles and Implementation
Algorithm Fundamentals and Mechanics
Differential Evolution is a population-based stochastic optimization algorithm belonging to the class of evolutionary computation methods. Unlike traditional gradient-based optimization techniques that require differentiable objective functions, DE operates effectively on non-linear, non-differentiable, and multimodal search spaces, making it particularly suitable for complex SPR sensor design optimization where the relationship between structural parameters and sensor performance is highly complex and interdependent [89] [88].
The algorithm maintains a population of candidate solutions (individuals) represented as real-valued vectors, with each vector element corresponding to an optimized parameter such as layer thickness, incident angle, or material composition. DE evolves the population through iterative cycles of mutation, crossover, and selection operations. The distinctive feature of DE lies in its mutation strategy, which generates donor vectors by calculating weighted differences between randomly selected population members according to the formula:
[ v{i,G+1} = x{r1,G} + F \cdot (x{r2,G} - x{r3,G}) ]
where (v{i,G+1}) represents the mutant vector, (x{r1,G}), (x{r2,G}), and (x{r3,G}) are distinct population vectors selected at random, and (F) is the mutation scale factor typically ranging between 0 and 1 [89] [88]. This differential mutation mechanism enables effective exploration of the search space while maintaining population diversity.
Following mutation, the crossover operation creates trial vectors by mixing parameters from mutant and target vectors based on a crossover probability ((Cr)). Finally, the selection process deterministically chooses between trial and target vectors for the next generation based on their fitness values, preserving only superior solutions. This evolutionary process continues until convergence criteria are met or a maximum number of generations is reached.
DE Variants for SPR Optimization
Several enhanced DE variants have been developed specifically to address challenges in SPR sensor optimization:
- Improved Differential Evolution (IDE): Incorporges adaptive parameter control and specialized boundary handling mechanisms to accelerate convergence while maintaining population diversity. In SPR biosensor optimization for waterborne bacteria detection, IDE achieved a sensitivity of 246.6°/RIU in just three iterations, significantly outperforming standard DE and FPS approaches [85] [88].
- Differential Evolution Particle Swarm Optimization (DEPSO): Hybridizes DE with Particle Swarm Optimization to combine DE's exploratory capabilities with PSO's social learning behavior. This hybrid approach has demonstrated remarkable success in phase-sensitive SPR gas sensor design, achieving phase sensitivity of (1.866 \times 10^6) deg/RIU while maintaining low reflectivity [89].
- Multi-objective DE: Extends the basic DE framework to simultaneously optimize multiple, often competing, performance objectives such as sensitivity, FOM, and detection accuracy through Pareto-based selection mechanisms [87] [86].
Figure 1: Differential Evolution Algorithm Workflow - The iterative process of population initialization, mutation, crossover, selection, and convergence checking.
DE-Optimized SPR Sensor Design: Methodologies and Protocols
Fitness Function Formulation for SPR Optimization
The cornerstone of effective DE-based SPR optimization lies in appropriate fitness function formulation, which quantitatively encodes design objectives into a scalar value that the algorithm seeks to maximize or minimize. The fitness function must comprehensively capture all critical aspects of sensor performance while accommodating practical fabrication constraints. For single-objective optimization targeting sensitivity enhancement, the fitness function ((F)) may be formulated as:
[ F = w1 \cdot S + w2 \cdot \left(\frac{1}{FWHM}\right) + w3 \cdot R{min} ]
where (S) represents sensitivity, (FWHM) is the full width at half maximum, (R{min}) denotes minimum reflectivity, and (w1), (w2), (w3) are weighting coefficients that prioritize different performance aspects [82] [88].
For multi-objective optimization addressing simultaneous enhancement of multiple performance metrics, the fitness function becomes more complex. Liu et al. [87] implemented a comprehensive multi-objective approach that concurrently optimized sensitivity (S), figure of merit (FOM), and depth-resolved figure of merit (DFOM) using the following formulation:
[ F{multi} = \alpha \cdot \frac{S}{S{ref}} + \beta \cdot \frac{FOM}{FOM{ref}} + \gamma \cdot \frac{DFOM}{DFOM{ref}} ]
where (S{ref}), (FOM{ref}), and (DFOM_{ref}) represent reference values from baseline sensor configurations, and (\alpha), (\beta), (\gamma) are application-specific weighting coefficients [87]. This approach achieved remarkable improvements of 230.22% in bulk refractive index sensitivity, 110.94% in FOM, and 90.85% in DFOM compared to conventional designs.
Implementation Protocol for DE-Based SPR Optimization
A systematic, step-by-step protocol for implementing DE to optimize multilayer SPR biosensors follows these key stages:
Problem Parameterization: Define the design variables to be optimized, which typically include metal layer thickness (Ag, Au), 2D material layer numbers or thicknesses (graphene, MXene, TMDCs), adhesive layer thickness (Cr, Ti), and incident angle. Establish practical constraints for each parameter based on fabrication limitations and physical realizability [82] [88].
Algorithm Configuration: Set DE control parameters including population size (NP), mutation factor (F), crossover rate (CR), and stopping criteria (maximum generations or convergence threshold). Typical values for SPR optimization problems range from NP=50-100, F=0.5-0.9, and CR=0.7-0.95 [85] [89].
Optical Modeling Integration: Implement the transfer matrix method (TMM) for multilayer optical systems to calculate reflectance spectra for each candidate solution. TMM computes the characteristic matrix for an N-layer structure as: [ M = \prod{m=1}^{N} Mm = \begin{bmatrix} M{11} & M{12} \ M{21} & M{22} \end{bmatrix} = \begin{bmatrix} \cos\betam & -\frac{i}{qm}\sin\betam \ -iqm\sin\betam & \cos\betam \end{bmatrix} ] where (\betam = \frac{2\pi dm}{\lambda} (\varepsilonm - n0^2\sin^2\theta0)^{1/2}) represents the phase shift, (dm) is layer thickness, (\varepsilonm) is dielectric constant, and (qm) denotes optical admittance [89] [86]. Validate optical modeling against finite element method (FEM) simulations where possible [82].
Iterative Optimization Execution: Execute the DE generational loop of mutation, crossover, and selection until convergence. Monitor population diversity and fitness progression to avoid premature convergence.
Solution Validation and Robustness Testing: Apply k-means clustering or similar techniques to identify robust design parameters from the optimized solution set that mitigate performance degradation from fabrication imperfections [87] [91].
Table 2: Experimental Protocols for DE-Optimized SPR Sensor Fabrication
| Stage | Protocol Description | Key Parameters | Validation Methods |
|---|---|---|---|
| Sensor Design | DE optimization of multilayer structure | Layer thicknesses, materials, incident angle | TMM, FEM simulation [82] |
| Substrate Preparation | Prism cleaning and surface functionalization | BK7/prism material, piranha treatment, oxygen plasma | Contact angle measurement |
| Metal Deposition | Thermal evaporation or sputtering of metal layers | Ag/Au thickness (40-70 nm), deposition rate | AFM, ellipsometry [88] |
| 2D Material Transfer | Wet/dry transfer of MXene, graphene monolayers | Layer number, transfer technique | Raman spectroscopy, SEM |
| Biofunctionalization | Immobilization of recognition elements | Antibody/aptamer concentration, incubation time | Fluorescence labeling, QCM |
| Performance Characterization | Angular/wavelength interrogation testing | Analyte concentration series, flow rate | Sensitivity, LOD, FOM calculation [87] |
Performance Analysis of DE-Optimized SPR Sensors
Enhanced Sensitivity and Detection Capabilities
DE-optimized SPR sensors consistently demonstrate remarkable improvements in sensitivity and detection limits across diverse applications. Bepare et al. [82] developed a DE-optimized SPR biosensor for Mycobacterium tuberculosis detection employing a CaF₂TiO₂/Ag/TiO₂/black phosphorus configuration that achieved exceptional angular sensitivity of 654 deg/RIU with a broad refractive index detection range (1.25-1.35). This performance represents a significant advancement over conventional SPR designs, enabling precise, label-free identification of diverse biological and chemical analytes.
In the domain of single-molecule detection, Liu et al. [87] [91] implemented a multi-objective optimization approach that attained a bulk refractive index sensitivity of 24,482.86 nm/RIU with a detection limit as low as 54 ag/mL (0.36 aM) for mouse IgG, establishing unprecedented sensitivity for label-free SPR detection. The optimized sensor demonstrated a broad linear dynamic range spanning from femtograms per milliliter (fg/mL) to micrograms per milliliter (μg/mL), making it suitable for both trace-level detection and high-concentration quantification.
For environmental monitoring applications, DE-optimized SPR biosensors configured for waterborne bacteria detection have achieved sensitivities of 246.6°/RIU for Escherichia coli identification using an Ag-MXene-graphene affinity layer structure [85] [88]. The IDE algorithm accomplished this performance level in just three iterations, dramatically reducing optimization time compared to conventional FPS methods while simultaneously improving accuracy.
Comparative Performance Analysis
The performance advantages of DE-based optimization become particularly evident when compared with alternative optimization strategies and conventional design approaches. The following table summarizes key performance metrics achieved by DE-optimized SPR sensors across various applications and configurations:
Table 3: Performance Comparison of DE-Optimized SPR Sensors
| Sensor Configuration | Optimization Method | Sensitivity | FOM | Detection Limit | Reference |
|---|---|---|---|---|---|
| Prism-CaF₂TiO₂/Ag/TiO₂/BP | DE Algorithm | 654 deg/RIU | 176.9 RIU⁻¹ | - | [82] |
| Ag-MXene-Graphene-Affinity | Improved DE | 246.6 deg/RIU | - | Waterborne bacteria | [85] [88] |
| Mouse IgG Immunosensor | Multi-objective PSO | 24,482.86 nm/RIU | - | 54 ag/mL (0.36 aM) | [87] [91] |
| Ag-BlueP/WS₂-Ag-MXene Gas Sensor | DEPSO | 1.866×10⁶ deg/RIU (phase) | - | Various gases | [89] |
| Conventional Au-film SPR | Fixed Parameter Scan | ~200 deg/RIU | ~80 RIU⁻¹ | ~pM range | [85] [86] |
The comparative data clearly demonstrates that DE-optimized configurations consistently outperform conventionally designed SPR sensors across all key performance metrics. The DEPSO-hybridized approach for gas sensing applications achieved phase sensitivity approximately an order of magnitude higher than conventional designs, while the multi-objective optimization for biosensing reduced detection limits by several orders of magnitude compared to standard SPR platforms [87] [89].
The Scientist's Toolkit: Research Reagent Solutions
Successful implementation of DE-optimized SPR biosensors requires carefully selected materials and reagents that collectively enable both high-performance sensing and efficient optimization.
Table 4: Essential Research Reagents and Materials for DE-Optimized SPR
| Reagent/Material | Function in SPR Sensor | Specification Notes |
|---|---|---|
| BK7 Prism | Optical coupling component for SPR excitation | Refractive index: 1.515 at 632.8 nm [85] |
| Silver (Ag) / Gold (Au) | Plasmonic metal layer for SPR generation | Ag thickness: 50-65 nm; Au: 45-55 nm [82] [88] |
| MXene (Ti₃C₂Tₓ) | 2D enhancement material | Monolayer thickness: ~1 nm; enhances electric field [85] [88] |
| Graphene | 2D enhancement material | Monolayer thickness: ~0.34 nm; increases adsorption [85] |
| Black Phosphorus (BP) | 2D enhancement material | Anisotropic properties; enhances sensitivity [82] |
| Transition Metal Dichalcogenides (TMDCs) | 2D enhancement material | WS₂, MoS₂; monolayer thickness: 0.6-0.7 nm [89] |
| Chromium (Cr) | Adhesive layer between prism and metal | Thin layer (1-2 nm); improves adhesion [87] |
| Capture Probes | Molecular recognition elements | Antibodies, aptamers, DNA probes; target-specific |
Future Directions and Research Frontiers
The integration of differential evolution with SPR sensor design continues to evolve, with several emerging frontiers promising further advancements. Machine learning and explainable artificial intelligence (XAI) approaches are increasingly being combined with evolutionary algorithms to accelerate optimization processes and enhance interpretability. Khatun and Islam [92] demonstrated that ML regression techniques coupled with SHAP analysis could significantly reduce computational costs while identifying the most influential design parameters, achieving wavelength sensitivity of 125,000 nm/RIU in PCF-SPR biosensors.
Future research directions likely include multi-scale optimization frameworks combining nanoscale material properties with macroscopic sensor architecture, automated experimental validation systems for closed-loop optimization, and multi-physics approaches incorporating fluidic, optical, and biochemical domains. Additionally, the development of application-specific DE variants tailored to particular sensing challenges—such as point-of-care diagnostics, environmental monitoring, and single-molecule detection—will further enhance the capabilities of SPR sensing platforms [87] [92].
As SPR technology continues toward miniaturization, portability, and disposability, DE optimization will play an increasingly crucial role in balancing multiple competing design constraints while maximizing sensing performance. The integration of low-cost solid-state light sources, including laser diodes, LEDs, and even smartphone displays, with optimized SPR architectures presents particularly promising opportunities for developing affordable, high-performance sensing platforms accessible beyond traditional laboratory settings [84].
Differential Evolution has established itself as a powerful, efficient, and versatile optimization methodology for advancing Surface Plasmon Resonance sensor design beyond the limitations of conventional approaches. By systematically navigating complex, high-dimensional parameter spaces, DE algorithms enable the discovery of multilayer configurations that simultaneously enhance sensitivity, improve detection limits, and maintain practical fabricability. The continued refinement of DE strategies—including hybrid approaches, multi-objective formulations, and machine learning integration—promises to further accelerate the development of next-generation SPR sensors with transformative capabilities for biomedical research, clinical diagnostics, and environmental monitoring.
In the field of Surface Plasmon Resonance (SPR) research, the evaluation of sensor performance relies on three fundamental metrics: Sensitivity, Figure of Merit (FOM), and Limit of Detection (LOD). These parameters provide distinct yet complementary insights into the capabilities of an SPR biosensor. Sensitivity quantifies the magnitude of the sensor's output response to a change in the refractive index at the sensing interface. The Figure of Merit (FOM) combines sensitivity with the resonance feature's sharpness to provide a comprehensive measure of sensor quality, while the Limit of Detection (LOD) defines the smallest measurable quantity of an analyte that can be reliably distinguished from zero. Understanding the definitions, interrelationships, and methodological considerations for these metrics is crucial for researchers developing new SPR technologies, optimizing experimental protocols, and interpreting biosensing data for applications ranging from fundamental molecular interaction studies to clinical diagnostics and drug discovery.
The significance of these metrics extends beyond theoretical comparisons, as they directly influence the practical utility of SPR biosensors in real-world applications. For drug development professionals, these parameters determine the ability to detect weak binding interactions, characterize low-affinity drug candidates, and monitor biomarkers at clinically relevant concentrations. The ongoing innovation in SPR technology, including the incorporation of novel materials and sophisticated detection schemes, continually redefines the benchmarks for these performance metrics. This guide provides an in-depth technical examination of sensitivity, FOM, and LOD within the context of modern SPR research, offering both theoretical foundations and practical methodologies for their evaluation and interpretation.
Defining the Core Performance Metrics
Sensitivity
Sensitivity (S) represents the core responsiveness of an SPR biosensor to changes in the refractive index (RI) of the sensing medium near the metal-dielectric interface. When target analytes, such as proteins or nucleic acids, bind to the sensor surface, they induce a localized increase in the RI. The sensitivity quantifies how effectively the sensor transduces this biochemical event into a measurable optical signal. The specific definition of sensitivity varies depending on the interrogation method employed by the SPR instrument.
In angular interrogation systems, sensitivity is defined as the shift in the resonance angle (θ) per unit change in refractive index (RIU), expressed as S = Δθ/Δn (deg/RIU). For wavelength interrogation systems, sensitivity is defined as the shift in the resonance wavelength (λ) per unit change in refractive index, expressed as S = Δλ/Δn (nm/RIU). The selection of materials and structural configuration significantly impacts the theoretical sensitivity. For instance, a recently proposed sensor utilizing silicon dioxide (SiO₂) and barium titanate (BaTiO₃) demonstrated an angular sensitivity of 568 deg/RIU [93], while another design employing a ring resonator supercell achieved a spectral sensitivity of 913.51 nm/RIU in its second resonance mode [94].
It is critical to distinguish between bulk sensitivity (response to changes in the refractive index of the entire solution) and surface sensitivity (response to molecular binding events confined to the sensor surface). Surface sensitivity is influenced by the penetration depth of the evanescent field, which typically extends 100-300 nm from the sensor surface. Configurations that enhance bulk sensitivity may sometimes do so at the expense of surface sensitivity, as demonstrated by plasmon-waveguide resonance (PWR) sensors, which exhibit increased penetration depth but reduced surface sensitivity compared to conventional SPR [23].
Figure of Merit (FOM)
The Figure of Merit (FOM) provides a more comprehensive assessment of sensor performance by incorporating both the sensitivity and the sharpness of the resonance dip. A sharper resonance dip (narrower Full Width at Half Maximum, or FWHM) enables more precise determination of the resonance position, which is crucial for detecting small changes. The most conventional definition of FOM is the ratio of sensitivity to the FWHM of the resonance curve: FOM = S / FWHM (RIU⁻¹).
However, research has revealed that this traditional definition may not always accurately predict the experimental Limit of Detection (LOD). Consequently, more sophisticated definitions of FOM have been developed. These generalized FOM formulations may incorporate additional parameters such as the depth of the plasmonic intensity dip (Isp) and the intensity contrast (ΔI), and are derived using statistical approaches like principal component analysis (PCA) and machine learning (ML) models trained on noise-included simulations [95]. For example, a sensor designed for cancer cell detection achieved a remarkable FOM of 223.74 RIU⁻¹ for HeLa cervical cancer cells [96], while the supercell ring resonator sensor reported an FOM exceeding 70 RIU⁻¹ for its third resonance mode [94].
Limit of Detection (LOD)
The Limit of Detection (LOD) represents the lowest concentration or mass of an analyte that can be reliably detected by the biosensor. Unlike sensitivity and FOM, which are intrinsic properties of the sensor's optical response, LOD is profoundly influenced by experimental conditions, including the molecular weight of the analyte, binding affinity, surface coverage of capture molecules, and the noise characteristics of the detection system [97].
In analytical chemistry, a standard definition for LOD is three times the standard deviation of the background (blank) noise [97]. The LOD can be expressed in terms of molar concentration (e.g., nM) or surface mass coverage (e.g., pg/mm²). The relationship between the optical signal and mass coverage is often defined using Resonance Units (RU), where 1 RU = 1 pg/mm² [97]. For an SPR instrument with an angular sensitivity of 0.1 mDeg, this corresponds to a mass sensitivity of approximately 0.6 pg/mm² or 0.6 RU [97]. In practical applications, an SPR biosensor developed for detecting chloramphenicol in blood samples achieved an impressive LOD of 0.099 ± 0.023 ng/mL [98], demonstrating the technology's capability for sensitive therapeutic drug monitoring.
Table 1: Key Definitions and Expressions of SPR Performance Metrics
| Metric | Mathematical Expression | Common Units | Key Influencing Factors |
|---|---|---|---|
| Sensitivity (S) | S = Δθ/Δn (angular)S = Δλ/Δn (spectral) | deg/RIU, nm/RIU | Prism material, wavelength, metal film properties, sensor geometry |
| Figure of Merit (FOM) | FOM = S / FWHM (conventional)Generalized FOM (includes Isp, ΔI) | RIU⁻¹ | Sensitivity, FWHM, resonance depth, detection scheme, noise |
| Limit of Detection (LOD) | LOD = 3 × σ_background (definition)Depends on specific assay | nM, pg/mm² | Background noise, analyte properties, binding affinity, surface chemistry |
Methodologies for Experimental Determination
Measuring Sensitivity
The experimental determination of sensitivity requires exposing the sensor to standardized solutions with known, varying refractive indices and measuring the corresponding shifts in the resonance signal. A common protocol involves using ethanol-water mixtures of different concentrations to create a series of refractive index standards [23]. The sensor's response (angular shift Δθ or wavelength shift Δλ) is then plotted against the refractive index change (Δn), with the slope of this plot yielding the experimental sensitivity.
For surface sensitivity assessment relevant to molecular binding, researchers often employ well-characterized protein layers. A monolayer of Cytochrome c, for instance, produces an angular shift of approximately 0.5 Deg, corresponding to a mass coverage of about 3000 pg/mm² [97]. This established relationship allows researchers to convert angular shifts into surface mass coverage, providing a critical link between optical response and bound analyte mass. When comparing sensitivity specifications across different instruments, it is essential to account for differences in prism material (e.g., BK7 vs. SF10 glass) and excitation wavelength, as these factors significantly influence the measured angular shift for the same molecular binding event [97].
Calculating Figure of Merit
Determining the FOM requires two primary measurements: the sensitivity (S) and the Full Width at Half Maximum (FWHM) of the resonance dip. The FWHM is measured directly from the resonance curve by finding the angular or spectral width at the point halfway between the baseline reflectance and the minimum reflectance of the dip.
To address the limitations of conventional FOM, researchers have developed sophisticated protocols involving shot-noise models and Monte Carlo simulations. These methods simulate the random noise inherent in optical detection systems (a dominant noise source around the plasmonic angle) by adding randomized shot-noise to theoretical reflectance spectra [95]. The plasmonic dip position is then recovered from thousands of noise-added iterations using curve-fitting algorithms. The standard deviation in the determined resonance position across these iterations provides an estimate of the measurement precision, which can be related to a more accurate, generalized FOM that better predicts experimental LOD.
Determining Limit of Detection
The experimental determination of LOD requires a systematic approach to account for all sources of variability in the measurement system. The recommended protocol begins with measuring the sensor's response in a blank solution (containing no analyte) multiple times to establish the standard deviation (σ) of the background noise. The LOD is then calculated as 3 × σ.
For concentration-based LOD, a dose-response curve (response vs. analyte concentration) must be constructed. As demonstrated in the development of an SPR biosensor for chloramphenicol, this involves preparing a series of standard solutions across the expected concentration range (e.g., 0.1 to 100 ng/mL) [98]. The assay's precision (intra-day and inter-day), accuracy, matrix effects, and extraction recovery rate should be rigorously validated through methodological verification [98]. The minimum detectable concentration is also highly dependent on biological factors, such as the equilibrium dissociation constant (KD) and the surface coverage of the capture molecule. For instance, with an anti-PNA antibody coverage of 5 × 10⁻¹⁶ mol/mm² and a KD of 20 nM, the minimum detectable concentration for PNA at equilibrium would be approximately 0.5 nM for an instrument with 0.1 mDeg angular sensitivity [97].
Advanced Sensor Configurations and Performance Comparison
Recent advancements in SPR sensor design have focused on incorporating novel materials and complex nanostructures to enhance performance metrics. These innovations often combine multiple materials to leverage their unique properties, such as high dielectric constants, enhanced field confinement, and chemical stability.
Table 2: Performance of Advanced SPR Sensor Configurations
| Sensor Configuration | Sensitivity | Figure of Merit (FOM) | Application/Target |
|---|---|---|---|
| BK7/SiO₂/Cu/BaTiO₃ [93] | 568 deg/RIU | 134.75 RIU⁻¹ | General refractive index sensing |
| Ring Resonator Supercell (Mode 2) [94] | 913.51 nm/RIU | N/R | Multimodal antigen detection |
| Ring Resonator Supercell (Mode 3) [94] | N/R | >70 RIU⁻¹ | Specialized biomedical applications |
| BK7/ZnO/Ag/Si₃N₄/WS₂ [96] | 342.14 deg/RIU | 124.86 RIU⁻¹ | Blood cancer (Jurkat) cell detection |
| BK7/SiO₂/Cu/BaTiO₃ [93] | 371.22 deg/RIU | 223.74 RIU⁻¹ | Cervical cancer (HeLa) cell detection |
| Ag, BaTiO₃, WSe₂ Hybrid [96] | ~300 deg/RIU (est.) | N/R | Dengue virus detection |
The use of high-refractive-index dielectric layers like silicon dioxide (SiO₂) and barium titanate (BaTiO₃) has been shown to enhance the electromagnetic field at the sensing interface, leading to improved sensitivity [93] [99]. Similarly, incorporating two-dimensional (2D) materials such as transition metal dichalcogenides (TMDCs: WS₂, MoS₂) can significantly boost performance by enhancing light-matter interactions and providing a larger surface area for biomolecular immobilization [96]. The configuration BK7/ZnO/Ag/Si₃N₄/WS₂ has demonstrated superior sensitivity for detecting cancerous cells compared to other 2D material combinations [96].
Nanostructuring has emerged as another powerful strategy for performance enhancement. Supercell ring resonator arrays and other metamaterial designs can support multiple resonance modes, enabling multimodal detection and dramatically increased sensitivity, as evidenced by the 913.51 nm/RIU sensitivity reported for the second resonance mode of a ring resonator design [94]. These structural innovations, combined with advanced materials, continue to push the boundaries of what is achievable with SPR biosensing technology.
Interrelationships and Technical Considerations
Understanding the intricate relationships between sensitivity, FOM, and LOD is essential for meaningful interpretation of SPR performance data and optimal sensor design. These metrics do not operate in isolation; improvements in one parameter often involve trade-offs with others, requiring careful balancing based on the specific application requirements.
Diagram 1: Logical relationships between SPR performance metrics and influencing factors. FOM is determined by optical parameters (S and FWHM), while LOD depends on both optical performance and experimental/assay conditions.
A critical technical consideration is that higher sensitivity does not automatically guarantee a lower LOD. The LOD is ultimately determined by the signal-to-noise ratio (SNR), which depends on both the magnitude of the response (governed by sensitivity) and the level of system noise [97] [95]. A sensor with high sensitivity but excessive noise may yield a poorer LOD than a less sensitive but quieter system. This explains why the FOM, which incorporates the resonance width related to measurement precision, often correlates better with LOD than sensitivity alone.
The selection of operational wavelength presents a fundamental trade-off. While longer wavelengths (e.g., near-infrared) can increase the penetration depth of the evanescent field, this often comes at the expense of surface sensitivity [97] [23]. For instance, an SPR instrument using 635 nm light may produce a 0.75 deg shift for a protein binding layer, while another with similar angular sensitivity but using 890 nm light might show only a 0.2 deg shift for the same binding event [97]. This highlights the importance of matching the sensor's operational parameters to the specific sensing application, particularly when targeting small molecules or thin molecular layers where surface sensitivity is paramount.
The Scientist's Toolkit: Essential Research Reagents and Materials
Successful SPR experimentation requires careful selection of materials and reagents that optimize sensor performance and ensure reproducible results. The following table outlines key components used in advanced SPR biosensing.
Table 3: Essential Research Reagents and Materials for SPR Biosensing
| Category | Specific Examples | Function in SPR Experiment |
|---|---|---|
| Prism Materials | BK7 glass (n=1.515), SF10 glass (n=1.723) | Couples incident light to excite surface plasmons; higher index prisms can reduce angular shift but improve detection range [97]. |
| Plasmonic Metals | Gold (Au), Silver (Ag), Copper (Cu) | Supports surface plasmon waves; Au offers biocompatibility and stability, Ag provides sharper resonances, Cu is a lower-cost alternative [93] [96]. |
| Dielectric Enhancement Layers | Silicon Dioxide (SiO₂), Barium Titanate (BaTiO₃), Zinc Oxide (ZnO) | Enhances electromagnetic field at interface, protects metal layer, improves sensitivity and FOM [93] [99]. |
| 2D Materials | WS₂, MoS₂, Graphene, MXene | Increases surface area for binding, enhances field confinement, protects against oxidation, improves sensitivity for thin analyte layers [96]. |
| Capture Molecules | Antibodies, DNA strands, Streptavidin | Provides specific recognition for target analytes; immobilized on sensor surface to capture binding partners [98]. |
| Surface Chemistry Reagents | CM5 dextran chips, EDC/NHS crosslinkers, Ethanolamine | Facilitates covalent immobilization of capture molecules onto the sensor surface [98]. |
| Running Buffers | HBS-EP (10mM HEPES, 150mM NaCl, 3mM EDTA, 0.05% surfactant P20) | Maintains consistent pH and ionic strength; surfactants minimize non-specific binding [98]. |
| Calibration Standards | Ethanol-water mixtures, Salt solutions, Certified RI liquids | Creates known refractive index changes for sensitivity calibration and instrument performance validation [23]. |
The strategic combination of these materials enables the fabrication of high-performance SPR sensors. For instance, a sensor structure might employ a BK7 prism as the coupling element, a silver layer for efficient plasmon excitation, a thin SiO₂ layer for protection and sensitivity enhancement, and a WS₂ monolayer to increase binding capacity and signal response [96]. The selection of immobilization chemistry, whether carboxymethylated dextran (CM5) matrix or self-assembled monolayers, must be compatible with both the sensor surface and the biological recognition elements to ensure optimal assay performance.
Surface plasmon resonance (SPR) and localized surface plasmon resonance (LSPR) are advanced optical sensing technologies that have revolutionized label-free biomolecular interaction analysis. Both techniques originate from the excitation of plasmons—collective oscillations of free electrons at metal-dielectric interfaces—but differ fundamentally in their physical manifestations and sensing capabilities [6] [100]. SPR represents a propagating electromagnetic wave along a continuous metal film, while LSPR is a non-propagating resonance confined to nanostructured metal surfaces [101]. These fundamental differences dictate their respective applications in research and drug development, with SPR excelling in detailed kinetic analysis and LSPR offering advantages in miniaturization and point-of-care diagnostics [6] [102]. This technical guide provides a comprehensive comparative analysis of these two plasmonic sensing modalities, framed within the broader context of fundamental SPR research.
The historical development of these technologies reveals their complementary nature. The theoretical foundation for surface plasmons was established in the early 20th century, with the first SPR biosensor demonstrated in 1983 [100] [102]. LSPR gained significant research momentum later, facilitated by advancements in nanotechnology that enabled precise fabrication of metallic nanostructures [6]. Today, both techniques have become indispensable tools for investigating molecular interactions without requiring fluorescent or radioactive labels [102], providing critical insights for drug discovery, diagnostic development, and fundamental biological research.
Theoretical Foundations and Working Principles
Surface Plasmon Resonance (SPR) Physics
SPR occurs when incident polarized light, under specific conditions of angle and wavelength, couples with the free electrons at a continuous metal film (typically gold or silver) to generate surface plasmon polaritons (SPPs) [6]. These SPPs are propagating electromagnetic waves that travel along the metal-dielectric interface, with their electric field intensity decaying exponentially perpendicular to the surface [6]. The resonance condition is highly sensitive to changes in the refractive index within the evanescent field, which typically extends 100-300 nm from the metal surface [103]. This sensitivity forms the basis for SPR sensing, where binding events between surface-immobilized ligands and analytes in solution alter the local refractive index, causing measurable shifts in the resonance condition [6] [102].
The propagation constant of SPPs is described by the equation: [ k{sp} = \frac{2π}{λ} \sqrt{\frac{\varepsilonm\varepsilond}{\varepsilonm+\varepsilond}} ] where (k{sp}) is the wavevector of the surface plasmon, (λ) is the wavelength of incident light, and (\varepsilonm) and (\varepsilond) are the dielectric constants of the metal and dielectric medium, respectively [6]. For resonance to occur, the wavevector of the incident light must match this propagation constant, which is typically achieved using prism couplers in the Kretschmann configuration [104].
Localized Surface Plasmon Resonance (LSPR) Physics
In contrast to propagating SPR, LSPR involves the confinement of surface plasmons to metallic nanoparticles or nanostructures with dimensions smaller than the wavelength of incident light [101]. When light interacts with these nanostructures, the electric field causes the conduction electrons to oscillate coherently, creating a localized plasmon that does not propagate along the surface [102]. The resonance condition depends strongly on the size, shape, composition, and local dielectric environment of the nanostructures [6] [101].
The LSPR phenomenon produces enhanced local electromagnetic fields near the nanoparticle surface, with a much shorter evanescent field decay length (typically 10-30 nm) compared to SPR [101]. This shorter decay length makes LSPR particularly sensitive to binding events occurring close to the nanoparticle surface but less sensitive to bulk refractive index changes [101]. The resonance is observed as sharp absorption and scattering peaks in the visible and near-infrared spectrum, with the exact spectral position being highly dependent on the nanoparticle's properties and immediate environment [6] [102].
For spherical nanoparticles much smaller than the wavelength of light, the extinction cross-section can be described by Mie theory: [ \sigma{ext} = \frac{24π^2R^3\varepsilond^{3/2}}{λ} \frac{\varepsiloni}{(\varepsilonr + 2\varepsilond)^2 + \varepsiloni^2} ] where R is the nanoparticle radius, (\varepsilond) is the dielectric constant of the surrounding medium, and (\varepsilonr) and (\varepsilon_i) are the real and imaginary parts of the metal's dielectric function [101].
Figure 1: SPR working principle showing light coupling through a prism to a metal film, with detection of biomolecular interactions via resonance angle shifts.
Figure 2: LSPR working principle showing nanoparticle interaction with light, causing spectral shifts and visible color changes upon molecular binding.
Comparative Performance Characteristics
Sensitivity and Detection Limits
The sensitivity of SPR and LSPR sensors is quantified through their response to changes in the refractive index of the surrounding medium, typically expressed in refractive index units (RIU) [6]. SPR generally exhibits higher bulk sensitivity due to its longer propagation length and deeper evanescent field penetration [103]. In contrast, LSPR's sensitivity is highly dependent on nanoparticle characteristics, with sharp corners and edges producing stronger local field enhancements [101] [102].
Table 1: Performance Comparison of SPR and LSPR Sensing Platforms
| Parameter | SPR | LSPR |
|---|---|---|
| Field Penetration Depth | 100-300 nm [103] | 10-30 nm [101] |
| Refractive Index Sensitivity | Higher for bulk changes [6] | Lower for bulk, higher for local changes [101] |
| Typical Detection Limit | Sub-nanomolar for proteins [104] | Picomolar to nanomolar [102] |
| Bulk Response Interference | Significant, requires correction [103] | Less pronounced [101] |
| Kinetic Analysis Capability | Excellent for binding constants [103] | Limited for fast kinetics [6] |
| Multiplexing Potential | Moderate (imaging SPR) [104] | High (array-based) [105] |
| Instrument Footprint | Larger, more complex optics [6] | Compact, simpler optics [105] |
Sensing Applications and Practical Considerations
SPR sensors have demonstrated exceptional capabilities in detecting various analytes across medical diagnostics, environmental monitoring, and food safety. For virus detection, SPR has achieved impressive sensitivity, with examples including influenza virus detection at 30 PFU/mL and Ebola virus detection at 0.5 pg/mL [104]. The technique is particularly valuable in drug development for determining binding affinity and kinetics between drug candidates and their targets [103] [102].
LSPR sensors leverage the unique optical properties of noble metal nanoparticles for diverse sensing applications. Gold and silver nanoparticles exhibit distinct colors due to their LSPR properties, and any alteration in their plasmonic environment—such as nanoparticle aggregation or changes in the local dielectric medium—causes measurable shifts in the LSPR peak accompanied by visible color changes [102]. This phenomenon has been exploited for developing colorimetric LSPR biosensors that enable visual detection without sophisticated instrumentation [102].
Table 2: Representative Applications and Detection Performance
| Application Area | SPR Performance | LSPR Performance |
|---|---|---|
| Virus Detection | Influenza: 30 PFU/mL [104]Ebola: 0.5 pg/mL [104] | Not specified in results |
| Protein Detection | Lysozyme: KD = 200 μM [103] | Soluble VEGF receptors [102] |
| Small Molecule Detection | Sulfamethoxazole in milk [100] | Patulin mycotoxin [102]Antibiotics in milk [102] |
| Environmental Monitoring | As(III) detection: 1.0 nM [102] | Organic vapors [101] |
Experimental Protocols and Methodologies
Standard SPR Experimental Protocol
Sensor Chip Preparation: SPR chips are typically prepared by depositing approximately 2 nm of chromium (as an adhesion layer) followed by 50 nm of gold onto clean glass substrates using electron beam physical vapor deposition [103]. The optimal metal film thickness (∼50 nm) is crucial for achieving a narrow and deep SPR minimum [104] [103]. Prior to experiments, surfaces are rigorously cleaned with RCA solutions (NH₄OH:H₂O₂:H₂O = 1:1:5 at 75°C) and oxygen plasma treatment to ensure reproducible results [103].
Ligand Immobilization: The gold surface is functionalized with recognition elements specific to the target analyte. For protein studies, this may involve forming self-assembled monolayers (SAMs) of alkanethiols followed by covalent attachment of ligands through EDC/NHS chemistry [103]. Alternatively, pre-functionalized sensor chips with carboxymethylated dextran matrices are commercially available and widely used for biomolecular interaction studies [102].
SPR Measurement: Experiments are conducted using an SPR instrument (e.g., Biacore, SPR Navi) with precise temperature control (typically 25°C) [103]. The running buffer (e.g., phosphate-buffered saline) must be thoroughly degassed and filtered (0.22 μm) to prevent air bubble formation during measurements [106]. Analyte solutions are injected at constant flow rates (typically 20-30 μL/min) to ensure controlled mass transport to the sensor surface [103]. The resonance angle shift is monitored in real-time, producing a sensorgram that reflects binding events.
Bulk Response Correction: A critical step in SPR data analysis involves correcting for the "bulk response" caused by refractive index differences between the running buffer and analyte solutions [103]. This can be achieved using a dual-channel reference system or advanced correction methods that utilize the total internal reflection (TIR) angle response to accurately determine the bulk contribution without requiring a separate reference surface [103].
Standard LSPR Experimental Protocol
Nanoparticle Synthesis and Characterization: Noble metal nanoparticles (typically gold or silver) are synthesized using chemical reduction methods [101]. The size, shape, and distribution of nanoparticles are carefully controlled, as these parameters directly determine the LSPR spectral position and sensitivity [101]. Spherical nanoparticles are commonly produced via citrate reduction of chloroauric acid, while nanorods and other anisotropic structures require more complex synthesis approaches using shape-directing agents [101].
Substrate Functionalization: Nanoparticles are immobilized on solid substrates (glass, silicon) or maintained in colloidal suspension, depending on the measurement configuration [105]. Surface functionalization with specific receptors (antibodies, aptamers, DNA probes) is performed to enable selective target detection [102]. For colorimetric assays in colloidal solutions, functionalization must also ensure nanoparticle stability to prevent nonspecific aggregation.
LSPR Measurement: Transmission or extinction spectra of LSPR sensors are measured using spectrophotometers or custom hyperspectral imaging systems [105]. The system typically incorporates a broadband light source (e.g., laser-excited phosphor source or tungsten-halogen lamp) and a spectrometer for spectral analysis [105]. Measurements can be performed in either single-particle mode using darkfield microscopy or ensemble mode averaging the response of many nanoparticles [101].
Data Analysis: The LSPR spectral peak position is tracked before and after exposure to the analyte. Binding events are quantified by measuring the wavelength shift (Δλmax) of the extinction maximum [101]. For colorimetric detection, the color change can be qualitatively assessed visually or quantitatively analyzed using RGB values from digital images [102].
Research Reagent Solutions and Essential Materials
Table 3: Essential Research Reagents and Materials for Plasmonic Sensing
| Item | Function | Specifications |
|---|---|---|
| Gold Sensor Chips | SPR substrate | 50 nm Au with 2 nm Cr adhesion layer [103] |
| Silver Nanoparticles | LSPR substrate | Spherical, 20-100 nm diameter [101] |
| Gold Nanorods | LSPR substrate | Aspect ratio 2-4 for NIR applications [101] |
| Thiol-Terminated PEG | Anti-fouling coating | MW 20 kDa, forms protein-resistant layer [103] |
| EDC/NHS Chemistry | Surface functionalization | Carbodiimide crosslinking for ligand immobilization [102] |
| PBS Buffer | Running buffer | 137 mM NaCl, 10 mM Na₂HPO₄, 2.7 mM KCl, pH 7.4 [103] |
| Prism Couplers | Optical components | Kretschmann configuration for SPR [104] |
Current Challenges and Emerging Solutions
Technical Limitations and Addressing Strategies
A significant challenge in SPR sensing is the bulk response effect, where molecules in solution that do not bind to the surface still generate signals due to the extended evanescent field [103]. This effect complicates data interpretation, particularly for complex samples or high analyte concentrations. Recent approaches to address this limitation include real-time bulk correction methods (e.g., PureKinetics by BioNavis) that measure the bulk refractive index directly during experiments [103] [106]. Additionally, proper experimental design with careful buffer matching and reference subtraction strategies can minimize bulk response artifacts [106].
LSPR sensors face challenges related to stability, reproducibility, and cost-efficient fabrication [102]. The sensitivity of LSPR is highly dependent on nanoparticle morphology, making reproducible fabrication essential. Advanced nanofabrication techniques such as electron-beam lithography and template-assisted methods have improved consistency in nanoparticle production [101]. For LSPR sensors in complex media, non-specific binding remains problematic, necessitating effective surface passivation using PEG coatings or other antifouling strategies [103].
Emerging Trends and Future Perspectives
The integration of SPR and LSPR with microfluidics represents a major advancement, enabling automated sample handling, reduced reagent consumption, and improved analysis throughput [6] [105]. Hyperspectral imaging systems for LSPR allow simultaneous characterization of multiple sensing spots, facilitating high-throughput screening applications [105]. These systems incorporate digitally controlled monochromators and automated data processing to streamline the optimization of sensor design parameters [105].
Hybrid plasmonic nanostructures combining different metallic elements or integrating plasmonic materials with other functional components show promise for enhancing sensor performance [102]. Bimetallic nanoparticles (e.g., gold-silver core-shell structures) enable fine-tuning of LSPR properties and improved stability [6]. The development of fiber-optic SPR and LSPR platforms offers opportunities for miniaturization and remote sensing applications, potentially expanding the use of plasmonic sensors beyond traditional laboratory settings [6] [100].
SPR and LSPR represent complementary rather than competing technologies in the landscape of plasmonic sensing. SPR remains the gold standard for detailed biomolecular interaction analysis, providing comprehensive kinetic information and higher sensitivity for bulk refractive index changes. Its well-established protocols and commercial instrumentation make it ideal for fundamental binding studies in drug development. Conversely, LSPR offers advantages in miniaturization, multiplexing, and point-of-care applications, with simpler optical systems and sensitivity to local binding events near nanoparticle surfaces.
The future of plasmonic sensing lies in leveraging the strengths of both technologies while addressing their respective limitations through continued materials innovation and instrumental advancements. As nanofabrication techniques improve and our understanding of plasmonic phenomena deepens, both SPR and LSPR will play increasingly important roles in biomedical research, clinical diagnostics, and drug discovery, providing researchers with powerful tools for investigating molecular interactions with unprecedented sensitivity and specificity.
Surface Plasmon Resonance (SPR) has evolved from a specialized laboratory technique to a cornerstone technology in pharmaceutical analysis, particularly in high-throughput screening (HTS). As an optical sensing technique, SPR enables real-time, label-free monitoring of biomolecular interactions by detecting changes in the refractive index at a sensor surface [16] [7]. This capability provides significant advantages over traditional endpoint assays, which risk false-negative results for interactions with fast dissociation kinetics and offer limited insight into the nature of molecular interactions [26]. The integration of SPR into HTS workflows represents a paradigm shift in drug discovery, allowing researchers to not only identify binding events but also characterize kinetic parameters and binding affinity simultaneously across thousands of candidate molecules [107] [20].
The fundamentals of SPR research are grounded in the Kretschmann configuration, where a light source interacts with a thin gold film to generate an evanescent wave that is exquisitely sensitive to changes in the surface environment [7]. When molecular binding occurs on the sensor surface, it alters the refractive index, resulting in a detectable shift in the resonance angle [20] [7]. This physical phenomenon forms the basis for SPR's application in studying diverse molecular interactions, from antibody-antigen binding to small molecule-target interactions, making it indispensable throughout the drug development pipeline [16].
Technological Advancements Enabling High-Throughput SPR
From Low-Throughput to High-Throughput Platforms
Traditional SPR systems were limited in throughput, analyzing interactions sequentially. The advent of SPR imaging (SPRi) and array-based systems has revolutionized this landscape, enabling simultaneous screening of hundreds or even thousands of interactions [107]. Modern high-throughput SPR (HT-SPR) instruments, such as the Carterra LSA platform, can feature 432 interaction spots simultaneously, generating kinetic data for entire antibody libraries in a single run [20]. This represents a significant advancement, with one study highlighting a ~2.2-fold increase from standard 384 commercial instrument capacity to approximately 864 protein ligand spots using Sensor-Integrated Proteome on Chip (SPOC) technology [26].
The core innovation enabling this throughput is multiplexed microfluidics integrated with sophisticated optics. These systems utilize sophisticated microfluidics that precisely deliver analytes over immobilized ligand arrays while CCD cameras monitor SPR dip profiles across all spots in real-time [20]. This parallel processing capability dramatically accelerates the screening timeline, as demonstrated by the collaboration between Eli Lilly and AbCellera, which identified a COVID-19 therapeutic antibody and moved it into clinical trials within just 90 days using HT-SPR technology [20].
Enhanced Sensitivity and Miniaturization
Continuous innovation in sensor design, surface chemistry, and optical components has significantly enhanced SPR's detection sensitivity [107]. Advances in nanomaterials and surface chemistry are enabling detection of lower analyte concentrations with higher specificity, while the emergence of portable and benchtop SPR systems is making the technology more accessible to non-specialist users [108]. The development of specialized sensor chips with various surface functionalities has further expanded application possibilities, allowing optimal immobilization strategies for diverse biomolecules from antibodies to membrane proteins [107].
Table 1: Key Technological Advancements in HT-SPR
| Advancement Area | Specific Innovation | Impact on HT-SPR |
|---|---|---|
| Throughput Capacity | Array-based SPR imaging (SPRi) | Simultaneous analysis of 384-864 interactions in a single run [26] [20] |
| Fluidics Technology | Multiplexed microfluidics | Parallel processing of multiple samples; reduced analysis time [107] [20] |
| Detection Sensitivity | Enhanced optics & nanomaterials | Lower limit of detection; ability to study weaker interactions [107] [108] |
| System Accessibility | Portable & benchtop systems | Expanded adoption beyond specialized core facilities [107] [108] |
SPR in High-Throughput Screening: Methodologies and Applications
Kinetic Characterization in Primary Screening
Traditional HTS approaches typically focus solely on identifying binders, often using endpoint assays that provide limited information about binding quality. HT-SPR transforms this paradigm by incorporating kinetic characterization directly into primary screening. This enables researchers to not only identify hits but also prioritize them based on association rates (kₐ), dissociation rates (kₑ), and affinity (K_D) from the earliest stages of discovery [20].
The significance of kinetic parameters extends beyond simple affinity measurements. A rapid association rate (kₐ) can lead to a quicker onset of pharmacological effect, while a slow dissociation rate (kₑ) translates to longer target residence time, potentially enhancing drug durability and allowing for less frequent dosing [20]. This detailed kinetic profiling is particularly valuable for therapeutic modalities like chimeric antigen receptor T-cell (CAR-T) therapies and antibody-drug conjugates (ADCs), where moderate affinity (K_D = ~50.0-100 nM range) has been found to correlate with improved clinical efficacy and reduced toxicity [26].
Experimental Protocol for HT-SPR Screening
A robust HT-SPR screening workflow involves multiple carefully optimized steps:
Surface Preparation: Sensor chips are functionalized with appropriate capture surfaces. Common approaches include carboxymethylated dextran hydrogels modified with anti-capture antibodies (e.g., anti-Fc for antibodies) or specific affinity tags (e.g., HaloTag, Strep-Tag) [26] [20].
Ligand Immobilization: The target molecule (ligand) is immobilized onto the sensor surface at densities optimized for kinetic analysis. Optimal density ensures sufficient signal while minimizing mass transport effects and steric hindrance. For HT-SPR, this process is parallelized across hundreds of spots simultaneously [20].
Analyte Injection: Compound libraries or analyte samples are injected over the sensor surface using precision microfluidics. Multi-concentration injections (e.g., 3-5 concentrations in a 3-fold dilution series) are typically used for full kinetic characterization [20].
Real-Time Monitoring: The association phase is monitored during sample injection, followed by dissociation phase monitoring during buffer flow. The entire interaction is tracked in real-time across all array spots [20].
Data Analysis: Sensorgram data is processed using specialized software to extract kinetic and affinity parameters. Global fitting algorithms simultaneously analyze data across all concentrations to determine kₐ, kₑ, and K_D values [20].
The following workflow diagram illustrates a specific HT-SPR application in immunomodulatory drug discovery:
Diagram 1: SPR-based screening workflow for CD28-targeted small molecules [109].
Essential Research Reagent Solutions
Successful implementation of HT-SPR relies on specialized reagents and materials optimized for label-free detection. The table below details key components:
Table 2: Essential Research Reagents and Materials for HT-SPR
| Reagent/Material | Function & Importance | Application Notes |
|---|---|---|
| SPR Sensor Chips | Solid supports with gold film and specialized coatings that enable biomolecular immobilization and signal detection [107]. | Available with various functionalities (carboxymethyl dextran, nitrilotriacetic acid (NTA) for His-tagged proteins, hydrophobic surfaces for membrane proteins) [107]. |
| Capture Reagents | Antibodies or affinity proteins that specifically capture target molecules in defined orientation [26]. | Anti-species Fc (e.g., anti-human Fc for human IgG), anti-tag antibodies (e.g., anti-HaloTag), or streptavidin for biotinylated ligands [26] [20]. |
| Running Buffers | Buffer systems that maintain biomolecular activity and minimize non-specific binding during analysis [107]. | Typically HEPES- or phosphate-buffered saline with added surfactant (e.g., Tween-20) and carrier protein to reduce non-specific binding [107]. |
| Regeneration Solutions | Conditions that dissociate bound analytes without damaging immobilized ligands for surface reuse [20]. | Low pH (glycine-HCl), high salt, or mild detergents; must be optimized for each specific interaction to maintain ligand activity over multiple cycles [20]. |
Addressing Drug Discovery Challenges with HT-SPR
Off-Target Binding Screening
Off-target binding is a major cause of adverse drug reactions and late-stage failures in drug development. SPR's real-time detection capability makes it uniquely suited for identifying transient off-target interactions that might be missed by traditional endpoint assays [26]. This is particularly critical since an estimated 33% of antibody candidates and numerous small molecule drugs exhibit off-target binding, contributing to approximately 30% of drug failures [26].
The SPOC (Sensor-Integrated Proteome on Chip) technology exemplifies how HT-SPR is advancing off-target screening. This next-generation platform enables high-density protein production directly onto SPR biosensors using cell-free expression systems, creating cost-efficient arrays for comprehensive off-target profiling against hundreds of potential interactors simultaneously [26]. This approach significantly reduces the risk of false negatives that can occur with traditional methods when investigating interactions with fast dissociation rates [26].
Affinity Optimization for Novel Therapeutic Modalities
HT-SPR provides critical insights for engineering optimal binding characteristics in emerging therapeutic classes. Contrary to conventional wisdom that higher affinity always equals better drugs, many modern modalities require precise affinity tuning for optimal efficacy [26]:
- CAR-T therapies: Moderate affinity (K_D = ~50.0-100 nM range) correlates with enhanced antitumor efficacy [26]
- Antibody-drug conjugates (ADCs): Reduced target affinity can improve tumor penetration and reduce on-target, off-site toxicity [26]
- Targeted protein degradation: Precise affinity balance is crucial for productive ternary complex formation and avoiding the "hook effect" [26]
HT-SPR enables researchers to rapidly screen and characterize thousands of variants to identify candidates with the optimal kinetic profile for each specific therapeutic context.
Future Perspectives and Market Outlook
Emerging Technological Trends
The SPR market is undergoing a transformative phase, fueled by several key technological trends. Hybrid systems that combine SPR with mass spectrometry and electrochemical detection are enabling more comprehensive molecular analysis in single platforms [107]. The integration of artificial intelligence (AI) and machine learning for data analysis is automating assay optimization and enhancing kinetic modeling, further accelerating screening throughput and data quality [107].
Miniaturization continues to advance, with portable SPR systems expanding applications into point-of-care diagnostics and resource-limited settings [107]. Additionally, sustainability-driven innovation is prompting the development of recyclable sensor chips and energy-efficient instrument designs, reducing the environmental footprint of pharmaceutical research [107].
Market Growth and Regional Adoption
The global SPR market demonstrates strong growth dynamics, valued at approximately $8.17 billion in 2025 and projected to advance at a CAGR of 14.44% through 2033, reaching $18.35 billion [108]. This expansion is driven by increasing adoption across industrial, commercial, and technological segments, particularly in pharmaceutical and biotechnology applications [107] [108].
Table 3: Surface Plasmon Resonance Market Outlook
| Region | Market Position & Characteristics | Key Growth Drivers |
|---|---|---|
| North America | Largest market share [107] | Presence of leading instrument manufacturers, advanced R&D infrastructure, high biopharmaceutical research investment [107] |
| Europe | Significant market share [107] | Strong pharmaceutical manufacturing base (Germany, U.K., France), government-funded research programs [107] |
| Asia Pacific | Fastest-growing region [107] [108] | Rapid biotechnology sector expansion, increasing healthcare investments, rising number of CROs, government innovation support [107] |
| Latin America, Middle East & Africa | Emerging markets [107] | Gradual adoption as bioscience research capacity strengthens, improving economic conditions [107] |
The following diagram illustrates the interconnected technological and market forces shaping the future of HT-SPR:
Diagram 2: Interconnected ecosystem driving HT-SPR adoption.
Surface Plasmon Resonance has firmly established itself as an indispensable technology in the future of pharmaceutical analysis, particularly through its integration into high-throughput screening paradigms. The unique ability of HT-SPR to provide real-time, label-free kinetic data across thousands of interactions simultaneously addresses critical challenges in modern drug discovery, from identifying specific binders to characterizing off-target interactions and optimizing therapeutic properties. As technological innovations in multiplexing, sensitivity, and data analytics continue to advance, coupled with expanding market adoption across global regions, SPR's role in accelerating and de-risking drug development is poised for continued growth. The ongoing transformation of SPR from a specialized characterization tool to a central platform in high-throughput screening workflows represents a fundamental advancement in pharmaceutical analysis, enabling more efficient discovery of safer and more effective therapeutics.
Conclusion
Surface Plasmon Resonance stands as an indispensable technology in modern biosciences, uniquely enabling the real-time, label-free quantification of biomolecular interactions. From elucidating fundamental kinetic parameters in drug discovery to enabling the rapid, sensitive detection of pathogens like Mycobacterium tuberculosis, SPR's versatility is unmatched. The ongoing integration of novel nanomaterials and sophisticated computational optimization promises a new era of even higher sensitivity and miniaturized, point-of-care diagnostic devices. As these advancements mature, SPR is poised to further revolutionize biomedical research and clinical diagnostics, solidifying its role as a cornerstone analytical technique for years to come.
References
- 1. The Basics of Label-free Biomolecular Interactions [https://www.horiba.com/usa/scientific/technologies/surface-plasmon-resonance-imaging/the-basics-of-label-free-biomolecular-interactions/]
- 2. Surface plasmon resonance [https://en.wikipedia.org/wiki/Surface_plasmon_resonance]
- 3. Surface Plasmon Resonance Biosensors: A Review of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10886473/]
- 4. Surface Plasmon Resonance (SPR) [https://cmi.hms.harvard.edu/surface-plasmon-resonance]
- 5. Latest surface plasmon resonance advances for G protein- ... [https://www.sciencedirect.com/science/article/pii/S2095177925001984]
- 6. Advancements in surface plasmon resonance sensors for real ... [https://pubs.rsc.org/en/content/articlehtml/2025/tc/d4tc04890c]
- 7. Surface Plasmon Resonance: An Introduction to a ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3045209/]
- 8. Configurations [https://www.sprpages.nl/spr-overview/configurations]
- 9. Kretschmann configuration based SPR sensor for the ... [https://www.sciencedirect.com/science/article/abs/pii/S0026265X25017059]
- 10. A comparative study of different types of sandwiched ... [https://www.sciencedirect.com/science/article/abs/pii/S1569441021000912]
- 11. Quantitative Cross-Platform Performance Comparison ... [https://www.mdpi.com/1424-8220/18/9/3136]
- 12. Experimental demonstration of lossy mode and surface ... [https://opg.optica.org/abstract.cfm?uri=ol-40-20-4739]
- 13. Evanescent wave-based optical biosensors for innovations ... [https://www.sciencedirect.com/science/article/pii/S2090123225005053]
- 14. Surface Plasmon Resonance Aptasensors: Emerging ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12190323/]
- 15. Surface plasmon resonance biosensors and their medical ... [https://www.sciencedirect.com/science/article/pii/S0956566325001824]
- 16. Optimizing drug discovery: Surface plasmon resonance ... [https://www.sciencedirect.com/science/article/pii/S2667022423002530]
- 17. Surface plasmon resonance - Latest research and news [https://www.nature.com/subjects/surface-plasmon-resonance]
- 18. SPR Sensorgram Explained [https://www.affiniteinstruments.com/post/spr-sensorgram-explained]
- 19. Surface Plasmon Resonance Sensorgram: A Step-by-Step ... [https://www.iaanalysis.com/surface-plasmon-resonance-sensorgram-guide.html]
- 20. SPR Essentials & Principles of High Throughput Kinetic ... [https://carterra-bio.com/resources/surface-plasmon-resonance-spr-essentials-principles-of-high-throughput-kinetic-analysis-march-19-2021/]
- 21. Use of Surface Plasmon Resonance (SPR) to Determine ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8489108/]
- 22. A Surface Plasmon Resonance-Based Integrated Assay for ... [https://pubmed.ncbi.nlm.nih.gov/40616443/]
- 23. Sensitivity Comparison of Surface Plasmon Resonance ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3111218/]
- 24. Surface plasmon resonance technology: Recent advances ... [https://www.sciencedirect.com/science/article/pii/S0165993623001668]
- 25. A Powerful Tool for Protein ... - Surface Plasmon Resonance [https://www.reichertspr.com/insights/blog-posts/surface-plasmon-resonance-a-powerful-tool-for-protein-protein-interaction-studies]
- 26. Real-Time SPR Biosensing to Detect and Characterize ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12190930/]
- 27. Comparison of methods for quantitative biomolecular ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8748344/]
- 28. Review Surface plasmon resonance biosensor chips [https://www.sciencedirect.com/science/article/abs/pii/S0731708525003590]
- 29. Surface Plasmon Resonance for Protein-Protein Interactions [https://www.affiniteinstruments.com/post/surface-plasmon-resonance-for-protein-protein-interactions]
- 30. Principle and Protocol of Surface Plasmon Resonance ( SPR ) [https://www.creativebiomart.net/resource/principle-protocol-principle-and-protocol-of-surface-plasmon-resonance-spr-361.htm]
- 31. CM-series [https://www.sprpages.nl/sensor-chips/biacore/cm-series]
- 32. Reichert Sensor Chip Buyer's Guide [https://www.reichertspr.com/insights/blog-posts/reichert-sensor-chip-buyers-guide]
- 33. -sensorchip SA [https://www.sprpages.nl/sensor-chips/biacore/sa]
- 34. Application Note 123: Surface Plasmon Resonance Measurement of... [https://biosensingusa.com/application-notes/application-note-123-surface-plasmon-resonance-measurement-protein-peptide-interaction-using-streptavidin-sensor-chip/]
- 35. A comparison of binding surfaces for SPR biosensing ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3840496/]
- 36. Immobilization [https://www.sprpages.nl/immobilization]
- 37. Surface Plasmon Resonance Sensors: A Guide for Scientists [https://nicoyalife.com/blog/spr-sensor-guide/]
- 38. 5 – Immobilization - Bruker Daltonics SPR [https://support.brukerspr.com/5-immobilization/]
- 39. A capture coupling method for the covalent immobilization ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3031178/]
- 40. Surface Plasmon Resonance SPR Market Report 2025 ... [https://www.cognitivemarketresearch.com/surface-plasmon-resonance-spr-market-report]
- 41. Surface Plasmon Resonance (SPR) Troubleshooting Guide [https://knowledge.kactusbio.com/trouble-shooting-guide-for-surface-plasmon-resonance-spr]
- 42. Top 10 tips for high quality SPR | data Guide SPR [https://nicoyalife.com/blog/spr-tips/top-10-tips-for-high-quality-spr-data/]
- 43. Using Surface Plasmon Resonance to Quantitatively ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5964981/]
- 44. Dissociation [https://www.sprpages.nl/kinetics/dissociation]
- 45. SPR-Measured Dissociation Kinetics of PROTAC Ternary ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6423499/]
- 46. Surface Plasmon Resonance (SPR) for the Binding ... [https://www.mdpi.com/1422-0067/26/8/3692]
- 47. QCM-D vs. SPR - What are the differences, and which ... [https://www.nanoscience.com/blogs/qcm-d-vs-spr-what-are-the-differences-and-which-technique-fits-your-application/]
- 48. Technical Guide: Expert Tips on Publishing Your SPR Data [https://nicoyalife.com/blog/publishing-spr-data/]
- 49. Theory and Applications of Surface Plasmon Resonance ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3230998/]
- 50. Fragment-Based Drug Discovery | April 15-16, 2025 [https://www.drugdiscoverychemistry.com/25/fragment-based-drug-discovery]
- 51. Fragment-based drug discovery: A graphical review [https://www.sciencedirect.com/science/article/pii/S2590257125000215]
- 52. Fragment-Based Drug Discovery (FBDD) [https://www.massbio.org/news/member-news/fragment-based-drug-discovery/]
- 53. Surface plasmon resonance-based biosensor for accurate ... [https://pubmed.ncbi.nlm.nih.gov/41186778/]
- 54. Detection of Mycobacterium tuberculosis complex and ... [https://www.sciencedirect.com/science/article/abs/pii/S0956566310003726]
- 55. Advancements in NIR sensing for tuberculosis detection ... [https://www.frontiersin.org/journals/sensors/articles/10.3389/fsens.2024.1521727/full]
- 56. Nanobodies: From Discovery to AI-Driven Design - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC12109276/]
- 57. Baseline drift [https://www.sprpages.nl/troubleshooting/baseline-drift]
- 58. Troubleshooting and Optimization Tips for SPR Experiments [https://www.creative-proteomics.com/resource/troubleshooting-optimization-tips-for-spr-experiments.htm]
- 59. Application of dynamic baseline adjustment based on ... [https://www.sciencedirect.com/science/article/abs/pii/S0030402622017284]
- 60. Data Processing of SPR Curve Data to Maximize the ... [https://www.mdpi.com/2079-6374/12/8/615]
- 61. Surface plasmon resonance as a high throughput method ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4656072/]
- 62. Analysis of binding kinetics and mass transport in SPR ... [https://www.sciencedirect.com/science/article/abs/pii/S0010482525014829]
- 63. 4 Ways to Reduce Non-Specific Binding in SPR Experiments [https://nicoyalife.com/blog/4-ways-reduce-non-specific-binding-spr/]
- 64. Suppressing Non-Specific Binding of Proteins onto ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6468902/]
- 65. Surface Plasmon Resonance Protocol & Troubleshooting [https://www.creativebiolabs.net/surface-plasmon-resonance.htm]
- 66. How Preconcentration can save your Protein Samples in ... [https://nicoyalife.com/blog/spr-tips/save-protein-samples-preconcentration-surface-plasmon-resonance/]
- 67. Blocking Strategies for IHC [https://www.thermofisher.com/us/en/home/life-science/protein-biology/protein-biology-learning-center/protein-biology-resource-library/pierce-protein-methods/blocking-strategies-ihc.html]
- 68. Regeneration [https://www.sprpages.nl/kinetics/regeneration]
- 69. Master the Art of Regeneration in SPR Experiments [https://nicoyalife.com/blog/regeneration-buffer-spr-experiment/]
- 70. Novel Regeneration Approach for Creating Reusable FO ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7828519/]
- 71. 2.5. Surface plasmon resonance (SPR) experiments [https://bio-protocol.org/exchange/minidetail?id=8583838&type=30]
- 72. The case for calibration-free concentration analysis - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC12203858/]
- 73. Two-dimensional nanomaterials as enhanced surface ... [https://www.sciencedirect.com/science/article/abs/pii/S0956566323006140]
- 74. Ultra-sensitive surface plasmon resonance sensor integrating ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12009283/]
- 75. A high-performance yet cost-effective Cu/Ag bimetallic SPR ... [https://www.sciencedirect.com/science/article/pii/S2214180425001503]
- 76. Advances in PCF-SPR sensors: a comprehensive review of ... [https://www.sciencedirect.com/science/article/abs/pii/S0263224125025850]
- 77. Editorial: 2D Materials for Advanced Sensors: Fabrication ... [https://www.mdpi.com/2079-4991/15/3/180]
- 78. An Enhanced Bimetallic Optical Fiber SPR Biosensor ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11902575/]
- 79. Experimental study of SPR sensor performance ... [https://www.sciencedirect.com/science/article/abs/pii/S1350449523004796]
- 80. Tailoring metallic layers to enhance the performance of ... [https://opg.optica.org/optcon/fulltext.cfm?uri=optcon-4-2-438]
- 81. Surface plasmon resonance biosensing [https://pubmed.ncbi.nlm.nih.gov/19151937/]
- 82. Optimized design of a surface plasmon resonance ... [https://pubmed.ncbi.nlm.nih.gov/41186760/]
- 83. Rapid identification of Mycobacterium tuberculosis ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3317816/]
- 84. Surface Plasmon Resonance Optical Sensor: A Review on ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6163427/]
- 85. Improved Differential Evolution Algorithm for Sensitivity ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10296569/]
- 86. Optimization methodology for structural multiparameter ... [https://www.sciencedirect.com/science/article/abs/pii/S0030401818308083]
- 87. Algorithm assisted comprehensive optimization of SPR ... [https://www.sciencedirect.com/science/article/abs/pii/S0956566325007225]
- 88. Improved Differential Evolution Algorithm for Sensitivity ... [https://www.mdpi.com/2079-6374/13/6/600]
- 89. Differential Evolution Particle Swarm Optimization for ... [https://www.mdpi.com/1424-8220/23/20/8401]
- 90. Optimized design of a surface plasmon resonance biosensor for ... [https://magazine.ospfound.org/2025/11/06/optimized-design-of-a-surface-plasmon-resonance-biosensor-for-mycobacterium-tuberculosis-detection-using-the-differential-evolution-algorithm/]
- 91. Algorithm assisted comprehensive optimization of SPR ... [https://pubmed.ncbi.nlm.nih.gov/40812130]
- 92. Design optimization of high-sensitivity PCF-SPR biosensor ... [https://journals.plos.org/plosone/article?id=10.1371/journal.pone.0330944]
- 93. Enhanced sensitivity and figure of merit in surface plasmon ... [https://www.sciencedirect.com/science/article/abs/pii/S0026265X25006964]
- 94. Supercell-enhanced multimodal plasmonic sensor for high- ... [https://www.nature.com/articles/s41598-025-08081-4]
- 95. Generalized figure of merit for plasmonic dip measurement ... [https://opg.optica.org/boe/abstract.cfm?uri=boe-13-4-1784]
- 96. On the sensing performance improvement in SPR ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12322250/]
- 97. Technical Note 102: SPR Sensitivity and Detection Limit [https://biosensingusa.com/technical-notes/technical-note-102-spr-sensitivity-detection-limit/]
- 98. Development of a surface plasmon resonance biosensor ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9805936/]
- 99. Figure-of-merit enhancement of surface plasmon ... [https://opg.optica.org/ol/abstract.cfm?uri=ol-37-7-1175]
- 100. Review of surface and plasmon ... resonance localized surface [https://link.springer.com/article/10.1007/s13320-011-0051-2]
- 101. Developments in Localized Surface Plasmon Resonance [https://link.springer.com/article/10.1007/s11468-024-02620-x]
- 102. Nanoplasmonic Biosensors: A Comprehensive Overview ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12067471/]
- 103. Accurate Correction of the “Bulk Response” in Surface ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9040059/]
- 104. ( Surface )- and Plasmon ... Resonance SPR Localized SPR [https://pmc.ncbi.nlm.nih.gov/articles/PMC8391291/]
- 105. Custom optical system for enhanced characterization and ... [https://www.sciencedirect.com/science/article/pii/S003040262500021X]
- 106. Bulk and Spikes [https://www.sprpages.nl/troubleshooting/bulk-and-spikes]
- 107. Surface Plasmon Resonance Market Size, Share, Trends ... [https://www.transparencymarketresearch.com/surface-plasmon-resonance-market.html]
- 108. Surface Plasmon Resonance (SPR) Market Transformation ... [https://www.linkedin.com/pulse/surface-plasmon-resonance-spr-market-transformation-lfvrf/]
- 109. Based Workflow for High-Throughput Discovery of CD28 ... [https://pubmed.ncbi.nlm.nih.gov/41244399/]